
用于药物发现的人工智能加速虚拟筛选平台 - Nature.com
Abstract
基于结构的虚拟筛选是早期药物发现的关键工具,人们对数十亿化合物库的筛选越来越感兴趣。然而,虚拟筛选的成功关键取决于计算对接预测的结合姿势和结合亲和力的准确性。在这里,我们开发了一种高精度的基于结构的虚拟屏幕方法 RosettaVS,用于预测对接姿势和结合亲和力。我们的方法在广泛的基准上优于其他最先进的方法,部分原因在于我们对受体灵活性进行建模的能力。我们将其纳入新的开源人工智能加速虚拟筛选平台中,用于药物发现。使用该平台,我们针对两个不相关的靶标(泛素连接酶靶标 KLHDC2 和人类电压门控钠通道 NaV1.7)筛选了数十亿个化合物库。对于这两个目标,我们发现了命中化合物,包括 KLHDC2 的 7 次命中(14% 命中率)和 NaV1.7 的 4 次命中(44% 命中率),所有化合物均具有个位数微摩尔结合亲和力。两种情况的筛查均在不到 7 天内完成。最后,高分辨率 X 射线晶体结构验证了 KLHDC2 配体复合物的预测对接姿势,证明了我们的方法在先导化合物发现中的有效性。
其他人正在查看类似内容
简介
基于结构的虚拟筛选通过识别有前途的化合物以供进一步开发和完善,在药物发现中发挥着关键作用。随着包含数十亿种化合物的易于访问的化学库的出现,人们对筛选广阔的化学空间以发现先导化合物越来越感兴趣。尽管筛选这些超大型文库有很多好处2,但只有少数成功的使用超大型文库的虚拟筛选活动被报道过3。此外,对于基于物理的对接方法来说,整个超大型文库的虚拟筛选变得越来越耗时且成本高昂。近年来,人们引入了多种技术来完成超大型文库虚拟筛选,包括开发可扩展的虚拟筛选平台以在高性能计算集群(HPC)4上并行对接运行、深度学习引导化学空间探索或主动学习技术仅筛选一小部分库以获得相似的性能5、6、7、8、基于分层结构的虚拟筛选9和GPU加速配体对接10。
但是,虚拟筛选的成功使用上述技术的筛选活动关键取决于配体对接程序的准确性,所述配体对接程序用于预测蛋白质-配体复合物结构以及区分和优先考虑真正的结合物与非结合物。领先的基于物理的配体对接程序,例如 Schrdinger Glide11、12、13、CCDC GOLD14 及其用于超大型文库筛选的虚拟筛选平台,并不是免费提供给研究人员的。Autodock Vina15作为广泛使用的免费程序之一,与Glide相比,其虚拟筛选精度略低。此外,缺乏一个开源的可扩展的虚拟筛选平台,该平台采用主动学习来实现超大型化学库虚拟筛选。深度学习技术的出现催生了许多模型16、17、18、19、20,旨在在显着缩短的时间内预测蛋白质-配体复杂结构。然而,这些方法更适合盲对接问题,其中小分子的结合位点未知。在结合位点已知的情况下(虚拟筛选中经常出现的情况),基于物理的配体对接方法继续优于深度学习模型21。此外,深度学习方法对于看不见的复合物的泛化性较差22。
在这项工作中,我们的目标是开发一种最先进的(SOTA)、基于物理的虚拟筛选方法和一种开放的方法。-源虚拟筛选平台,能够稳健、高效地筛选数十亿化合物库。这是通过改进我们之前用于虚拟筛选的基于物理的 Rosetta 通用力场 (RosettaGenFF)23 来实现的,产生一个名为 RosettaGenFF-VS 的改进力场。基于这个新的力场,我们使用 Rosetta GALigandDock23(以下简称 RosettaVS)开发了最先进的虚拟筛选协议。此外,我们采用了之前工作中的对接协议,以允许很大的受体灵活性,使我们能够在虚拟筛选协议中模拟灵活的侧链以及有限的骨干运动。事实证明,这对于某些需要对配体结合时诱导构象变化进行建模的靶标至关重要。然后,我们创建了一个高度可扩展的开源 AI 加速虚拟筛选平台 (OpenVS),集成了药物发现的所有必要组件(图 1a)。我们使用 OpenVS 平台针对两种不相关的蛋白质筛选数十亿个化合物库:KLHDC224,25、人类泛素连接酶和人类电压门控钠通道 NaV1.726。整个虚拟筛选过程在7天内在本地HPC集群上完成,该集群为每个目标配备3000个CPU和一个RTX2080 GPU。从最初的虚拟筛选活动中,我们发现了一种针对 KLHDC2 的化合物和四种针对 NaV1.7 的化合物,所有化合物均表现出个位数的 M 结合亲和力。将重点库与我们的虚拟筛选平台结合使用,又发现了六种与 KLHDC2 具有相似结合亲和力的化合物。最后,KLHDC2 复合物的对接结构通过 X 射线晶体学验证,显示与预测的结合姿势非常一致。这种探索、管理和测试的迭代过程强调了我们方法的稳健性及其在大型分子库中发现有前途的化合物的潜力。
结果
人工智能加速虚拟系统的开发筛选平台
我们之前开发的Rosetta GALigandDock是一种配体对接方法,它使用基于物理的力场RosettaGenFF,该方法之前在配体对接精度方面表现出了卓越的性能23。该方法可以对蛋白质-配体复合物进行精确建模,适应受体侧链的完全灵活性和主链的部分灵活性。然而,它并不直接适用于大规模虚拟筛选,因为:(a)它无法准确地模拟某些官能团(因为原始方法是在数十万种化合物上进行测试,而不是本研究中的数十亿种化合物);(b) 缺乏用于对与同一靶标结合的不同化合物进行准确排序的熵模型。此外,使用基于物理的虚拟筛选方法将每个单独的化合物对接在数十亿个化合物库中的成本极其昂贵。
为了解决这些问题,我们整合了多项增强功能并纠正了几个关键问题,以促进数十亿个小分子的建模。首先,我们通过合并新的原子类型和新的扭转势来改进 RosettaGenFF,并改进预处理脚本(参见方法)。其次,我们开发了用于虚拟筛选的 RosettaGenFF-VS,对与同一目标结合的不同配体进行排名,它将我们之前的模型焓计算 (H) 与估计配体结合时熵变 (S) 的新模型结合起来(详细信息请参阅方法).
为了能够筛选超大化合物库,我们采用了两种策略。首先,我们开发了一种改进的对接方案RosettaVS,它实现了两种高速配体对接模式:虚拟筛选快速(VSX)是专为快速初始筛选而设计的,而虚拟筛选高精度(VSH)是一种更准确的方法。用于初始屏幕中热门点击的最终排名。两种模式之间的主要区别在于 VSH 中包含完整的受体灵活性(更多详细信息,请参阅方法)。
即使有这些加速,对接超过 10 亿个化合物的成本也高得令人望而却步。基于最近的工作4、5、6、7、8,我们开发了一个开源虚拟筛选(OpenVS)平台,该平台使用主动学习技术在对接计算期间同时训练特定目标的神经网络,以有效地分类和选择最有希望用于昂贵的对接计算的化合物。该平台还被设计为针对大规模虚拟屏幕具有高度可扩展性和可并行性。
RosettaVS 在虚拟筛选基准测试中展示了最先进的性能
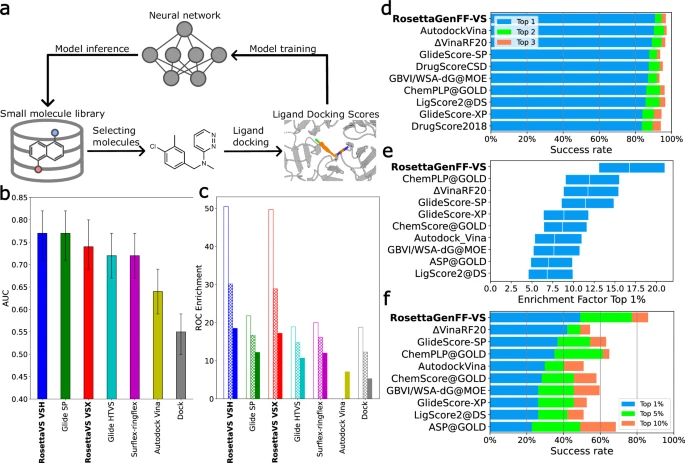
我们首先使用2016 年评分函数比较评估 (CASF2016) 数据集27,28 用于对 RosettaGenFF-VS 的性能进行基准测试。CASF2016 由 285 个不同的蛋白质-配体复合物组成,是专门为评分功能评估而设计的标准基准。它提供所有小分子结构作为诱饵,有效地将评分过程与分子对接固有的构象采样过程分离。我们用对接功率测试来衡量对接精度,用筛选功率来衡量筛选精度。基于深度学习的评分函数的最新发展已在这些基准上表现出卓越的性能16,20,29,但是,尚不清楚这些方法对于看不见的化合物和受体的通用性如何。此外,这些方法没有采用严格的训练/测试分割。即使使用配体 0.6 Tanimoto 相似性的截止值和蛋白质 30% 的序列同一性,这些验证基准的污染仍然可能发生。因此,我们后续的比较将集中在其他基于物理的评分函数,包括参考文献中表现最好的函数。28. 如图 1d 和补充图 3 所示,RosettaGenFF-VS 实现了准确地区分天然结合姿势与诱饵结构的领先性能。对结合漏斗的进一步分析(测量驱动构象采样向最低能量最小值的能量势效率)显示,RosettaGenFF-VS 在广泛的配体 RMSD 中具有优异的性能,这表明与相比,可以更有效地搜索最低能量最小值。到其他方法(补充图7)。接下来,进行筛选能力测试以评估评分函数在众多负小分子中识别真正结合物的能力。两个指标用于评估筛选能力测试中评分函数的性能。第一个指标是富集因子 (EF),它衡量对接计算在所有回收化合物的给定 X% 截止值下识别真阳性早期富集的能力(详细信息请参阅方法)。第二个指标是在数据集中 1%、5% 或 10% 顶级配体整体目标蛋白中放置最佳结合剂的成功率。在图 1e 和补充图 4 中,RosettaGenFF-VS 的前 1% 富集因子 (EF1% = 16.72) 明显优于第二好的方法 (EF1% = 11.9)。同样,图 1f 和补充图 5 表明,RosettaGenFF-VS 在识别排名前 1/5/10% 的分子中的最佳结合小分子方面表现出色,优于所有其他方法。对我们的方法对各种筛选能力子集28的分析显示,与其他方法相比,在更极性、更浅和更小的蛋白质口袋方面有显着改进(补充图8)。然而,在现实的虚拟筛选场景中,对接方法必须准确地对复合物进行评分,同时还要有效地对构象进行采样。
为此,我们进一步从RosettaVS协议中评估了VSX和VSH模式的虚拟筛选性能有用诱饵目录 (DUD) 数据集30。DUD 数据集由 40 个药物相关蛋白质靶标和超过 100,000 个小分子组成。AUC 和 ROC 富集这两个常见指标用于量化虚拟筛选性能。受试者工作特征 (ROC) 曲线已广泛用于评估虚拟筛选性能,其目的是区分活性化合物和诱饵化合物。ROC 曲线下面积 (AUC) 评估区分活性物质与诱饵的方法的整体性能。ROC 富集解决了富集因子31的一些缺陷,测量给定 X% 假阳性率下的真阳性富集。早期富集是大规模虚拟屏幕的关键因素,因为当前实验通量的限制通常只能合成和实验测试数十到数百种化合物。结果,就 AUC 和 ROC 富集而言,将 RosettaVS 定位为领先的虚拟筛选方法(图 1b、c 和补充图 9)。值得注意的是,RosettaVS 的性能比第二好的方法高出两倍(0.5/1.0% ROC 富集),在早期 ROC 富集方面实现了最先进的性能,进一步凸显了 RosettaVS 的有效性。此外,VSH 模式稍微优于 VSX 模式,因为它能够模拟配体诱导的口袋侧链的构象变化(参见方法)。
发现 KLHDC2 泛素连接酶的小分子命中
为了展示我们新开发方法的有效性,我们开始对人类 KLHDC2 泛素连接酶 24,25 进行大规模虚拟筛选,该酶尚未与任何已知的药物样小分子结合剂连接。作为 CUL2-RBX1 E3 复合物的底物受体亚基,KLHDC2 具有 KELCH 重复螺旋桨结构域,可以以纳摩尔亲和力识别其底物的二甘氨酸 C 端降解决定子。我们着手鉴定可以锚定到 KLHDC2 二甘氨酸结合位点的化合物,KLHDC2 最近被认为是一种有前途的 PROTACs E3 平台,用于靶向蛋白质降解32,33。
我们使用了 OpenVS 平台和 VSXRosettaVS 中的模式可针对目标蛋白结构筛选 Enamine-REAL 文库,该文库包含约 55 亿个可购买小分子,合成成功率为 80%(请参阅 AI 加速虚拟筛选的方法-工作流程)。如补充图 11 所示,该协议在每次对接迭代后发现了具有更高预测结合亲和力的更好化合物。前 0.1 个百分点的预测结合亲和力从第一次迭代中的 6.81 kcal/mol 显着提高到最终迭代中的 12.43 kcal/mol。当虚拟筛选达到最大第十次迭代时,我们停止了虚拟筛选,特别是因为在第八次迭代之后没有识别出新的全局最小结构。随后,我们使用RosettaVS中的VSH模式重新对接了虚拟筛选中排名靠前的50,000个小分子,从而在对接过程中实现了受体结构的灵活性。整个计算在一周内在配备 3000 个 CPU 和一个 RTX2080 GPU 的本地 HPC 集群上完成。Enamine REAL 库中约 600 万个化合物(0.11%)进行了对接。
我们选取了排名靠前的 1000 个化合物,并过滤掉了预测溶解度较低、结合构象中氢键不饱和的化合物,然后进行相似性聚类以减少配体结构的冗余。在 PyMol34 中手动检查了总共 54 个通过过滤和聚类的分子的有利相互作用和几何形状。最终选择了37个化合物进行化学合成。其中,成功合成了 29 种化合物(补充图 13),并在 AlphaLISA 竞争测定中进行了表征,其中测试了每种化合物与含有二甘氨酸的 SelK C 末端降解决定子肽竞争结合 KLHDC2 的能力。虽然几种化合物在取代降解决定子肽方面表现出可检测的活性,但化合物 29 (C29) 脱颖而出,其最佳 IC50 约为 3 M(图 2a、c)。这个个位数的 MIC50 在使用 BioLayer 干涉测量法的竞争测定中得到了进一步证实(补充图 14)。
为了揭示化合物 29 的结合模式,我们将 apo KLHDC2 晶体与该化合物浸泡并测定KLHDC2-C29 复合物的结构,分辨率为 2.035。与其置换二甘氨酸肽的活性一致,化合物 29 与降解决定子结合袋结合,其远端羧基与 KLHDC2 中参与识别降解决定子 24 的 C 末端(图 3)。该化合物羧基旁边的三唑部分位于三个芳香族残基(Tyr163、Trp191 和 Trp270)之间,并通过 NHN 氢键稳定。通过 Lys147 和小分子中心羰基之间形成的氢键以及叔丁基苯基直接堆积到降解决定子结合袋的辅助室,进一步增强了化合物与 E3 的相互作用。与化合物两端相比,化合物中间的二甲基硫醚连接体显示出较差的电子密度,表明结构灵活性较高(图2e)。总体而言,化合物 29 的结合模式与二甘氨酸 C 端降解决定子的结合模式高度相似,其结合姿势与预测非常匹配(图 2f)。
在我们最初的发现之后,我们扩大了我们的探索ZINC22 库36 包含约 41 亿个可直接对接 3D 格式的小分子。值得注意的是,ZINC22 的很大一部分来自 Enamine REAL 库。我们针对 ZINC22 文库对乙酰基-氨基-甲基-三唑-乙酸主链(图 2a、b 中以红色突出显示的二维结构)进行了子结构搜索,并鉴定了约 381,567 种化合物。这些化合物使用 GALigandDock 灵活对接模式进行对接,通过手动检查通过上述所有过滤器的前 100 个结构,挑选出 21 个化合物。合成了这些化合物(补充图 15),并在基于 AlphaLISA 的竞争测定中测试了它们的活性,并以化合物 C29 作为阳性对照。值得注意的是,另外六次命中显示出个位数的 M IC50,进一步验证了我们方法的有效性(图 2b、d)。未来需要优化以提高这些化合物的效力以达到纳摩尔范围。为了测试我们筛选程序的可靠性,我们重新进行了部分计算实验,并重新发现了已确认的最佳化合物 C2.8。(详情参见方法)
NaV1.7 VSD4 小分子拮抗剂的发现
为了评估我们的虚拟筛选方案的更广泛适用性,我们检查了其对人体电压的有效性 -门控钠通道,hNaV1.7。具体来说,我们靶向电压传感结构域 IV (VSD4),它参与 NaV 通道快速失活37,38,39,40,并包含稳定通道失活状态的小分子受体位点26,41,42。我们使用相同的虚拟筛选方案根据 ZINC22 库(约 41 亿种化合物)筛选靶标。与 KLHDC2 筛选类似,每次迭代后都会发现具有更好预测结合亲和力的新化合物,并且前 0.1 个百分点的预测结合亲和力从第一次迭代中的 10.8 kcal/mol 提高到最终迭代中的 18.2 kcal/mol。虚拟筛选在第七次迭代后停止,其中顶部预测的结合亲和力达到收敛(补充图12)。使用 RosettaVS 中的 VSH 模式对虚拟筛选中排名靠前的 100,000 个小分子进行重新对接,以考虑受体结构的灵活性。ZINC22 文库中约 450 万种化合物 (0.11%) 进行了对接。
我们首先对排名前 100,000 个的小分子进行聚类,然后对前 1000 个聚类代表分子应用过滤器。手动检查了总共 160 个通过聚类和过滤的分子。为了确保我们选择的化学新颖性,我们特别排除了含有已知芳基磺酰胺弹头或结构类似于抗组胺药或 β 受体阻滞剂的分子。最后,选择了 10 个与 ChEMBL 数据库 43,44 中已知的 NaV1.7 抑制剂的 Tanimoto 相似度小于 0.33 的分子进行合成。其中,9 个已成功合成(补充图 16),并使用全细胞膜片钳电生理学测定对 HEK-293 细胞中稳定表达的 hNaV1.7 通道进行测量,如方法中所述。化合物 Z8739902234 以灭活状态依赖性方式对 NaV1.7 表现出最高效力,IC50 = 1.3 M(图 4 和补充图 18)。四种化合物的 IC50 值优于 10 M,转化为 44.4% 的命中率(补充图 17)。值得注意的是,化合物 Z8739902234 具有状态依赖性(补充图 18,左图),并且相对于 hNaV1.5 和 hERG 通道,对 hNaV1.7 具有中等选择性(补充图 18,右图)。
讨论
在这项工作中,我们提出了一种最先进的基于物理的虚拟筛选方法,该方法集成到一个全面的可扩展平台中,该平台使用主动学习进行大规模虚拟筛选和先导化合物发现。我们的方法发现了新 E3 连接酶 KLHDC2 的 7 个结合物,以及人类电压门控钠通道 NaV1.7 VSD4 的 4 个结合物。
RosettaGenFF-VS 和 RosettaVS 在CASF2016 和 DUD 基准分别将其确立为领先的基于物理的配体对接和虚拟筛选方法。RosettaVS 的显着性能来自于两大进步。首先,高对接精度和采样效率的结合使得配备RosettaGenFF-VS的虚拟筛选方案能够比其他方法更有效地找到蛋白质-配体复合物的正确结合最小值。其次,与大多数其他虚拟筛选方法仅适用于疏水性更强、更深和更大的蛋白质袋不同,我们的方法还展示了具有更多极性、更浅和更小的袋的高性能,这可能是由于蛋白质的更好平衡-RosettaGenFF-VS 获得的配体与配体内分子能量。
尽管我们的方法在所有方面都优于现有方法,但我们相信还有进一步改进的空间。人工智能在各个科学领域的应用激增,包括蛋白质结构预测45,46、药物发现47,48和材料设计49,是近年来的一个显着趋势。我们协议的未来增强可能会涉及 GPU 加速和深度学习模型的集成,例如,使用 GPU 加速配体对接 10 或使用生成式 AI 进行高效姿势生成 50,51。其他潜在的改进包括完善替代主动学习模型,以更好地指导化学空间探索,并结合可推广的基于深度学习的评分函数,以提高对真实结合物的辨别力。我们方法的另一个改进是能够使用已知的非小分子结合物(例如大环化合物或抗体环)作为模板结构来指导小分子虚拟筛选。我们预计,基于结构的虚拟筛选与深度学习技术相结合的进一步发展将显着提高虚拟筛选活动的准确性和效率。
方法
计算方法
计算方法分为三个主要部分。第一部分重点介绍用于虚拟筛选的 Rosetta 通用力场 (RosettaGenFF-VS) 和 Rosetta 虚拟筛选协议 (RosettaVS) 的开发。第二部分介绍了 RosettaGenFF-VS 和 RosettaVS 的基准测试。最后一部分提供有关人工智能加速虚拟筛选协议的详细信息。
RosettaGenFF-VS 和 RosettaVS 的开发
在本节中,我们将介绍有关熵的发展的详细信息用于增强 Rosetta 通用力场的模型,以便对与同一目标结合的不同配体进行排序,以及我们在 Rosetta GALigandDock 中开发的 Rosetta 虚拟筛选协议。
熵估计
在虚拟中在筛选任务中,结合时冻结配体扭转和刚体自由度引起的熵对结合自由能的贡献是近似的。不考虑受体的贡献。结合时的熵变采用以下形式:
配体刚体自由度 (Srb) 的熵变近似为分子质量 (m)52 的函数。假设结合态的绝对熵可以忽略不计,则配体扭转(Storsion)的熵变直接根据其未结合态的熵估计。为了估计未结合状态下每个可旋转扭转值的概率分布,使用 RosettaGenFF 在室温 (300 K) 下收集每 3000 个仅配体蒙特卡罗 (MC) 模拟步骤的构象。然后将概率分布转换为熵:
其中 R 是气体常数,T 是 300 K,pij 是给定扭转角 i 位于 bin j 中的概率(bin 大小为 60)。因为我们假设配体扭转是独立的,所以配体扭转的净熵损失只是总体可旋转扭转的这些因素的总和。该方案有效地捕获了未结合状态下配体扭转角的预组织效应,因此解决了将这些角度在未结合时视为完全自由的更简单算法中的缺点。Srb 和 Storsion 的最佳权重是使用网格搜索来最大化 DUD-E 集 53 的非重叠子集的 AUROC 获得的(更多详细信息,请参阅 SI,DUD-E 的子集)。此搜索中考虑的权重 (w1, w2) 为 {(0,0), (0,1), (1,0), (1,1), (0,2), (2,0), (2,1), (1,2), (2,2)}。获得的最佳权重(刚体和扭转角分别为 2.0 和 1.0)与均匀权重的天真猜测(两者均为 1.0)相差不大。与未校正的结果相比,包括熵估计在内的 AUROC 指标提高了 3%,并且对基于富集的指标的影响可以忽略不计。这是默认的熵估计方法,因此我们将其称为默认熵。
还实现了第二种熵估计方法。我们将这种方法命名为“简单熵”,与第一种方法相比,它采用了更简单的公式。我们没有使用 MC 模拟,而是使用旋转键的数量来估计未结合小分子的扭转熵。尽管这种方法忽略了配体在未结合状态下的预组织效应,但我们使用这种更简单的方法观察到了可比较的基准结果。这种更简单方法的方程如下:
最佳权重是通过网格搜索确定的,范围从 0.0 到 3.0,步长为 0.1。这样做是为了最大限度地提高预测与源自 PDBbind54 精炼数据集的数据集的实验结合亲和力之间的相关性。这个精心策划的数据集称为 PDBbind-refine-no-metal 数据集,是通过排除复合物中存在金属离子的情况来准备的。为了防止数据泄露,CASF2016中的任何案例也从PDBbind-refine-no-metal数据集中删除。最佳权重为 w1 = 0.0 和 w2 = 0.4,表明分子量对熵估计没有贡献。因此,简单熵的最终公式为 \(\Delta S=0.4*{n}_{{rotor}}\)。
结合亲和力估计
结合亲和力为通过将焓和熵贡献相加来估计。使用以下方程估算结合时的焓变 (H):
其中 Ecomplex、E Protein、Eligand 分别是复合物、蛋白质和配体的 Rosetta 能量,来自 Rosetta 通用力场(RosettaGenFF)。RosettaGenFF 结合了 Rosetta 蛋白质能量模型55,56,57 和非蛋白质分子的通用能量术语,即 Lennard-Jones、库仑、氢键、隐式溶剂化和通用扭转能。RosettaGenFF 的完整描述可以在参考文献中找到。23 H本质上是来自RosettaGenFF的蛋白质和配体之间的相互作用能。然后使用方程计算预测的结合亲和力:\(\Delta G={dH}-{TdS}=\Delta H+\Delta S\),其中温度 T 隐式包含在上述 S 估计中。该估计的结合亲和力用于对与同一靶标结合的不同配体进行排序。值得注意的是,估计的结合亲和力的尺度是任意的,并且它并不直接对应于实验的结合亲和力。我们将这种熵增强力场命名为用于虚拟筛选的Rosetta通用力场(RosettaGenFF-VS)。
Rosetta虚拟筛选(RosettaVS)评估模式
中的评估模式RosettaVS 是为复杂结构的结合亲和力估计而开发的。它提供了多种用于估计结合亲和力的选项。它可以估计所提供结构的结合亲和力,在口袋内单独执行配体的局部最小化,或在配体的某个截止值(默认为 4.5 )内执行配体和一组口袋残基的局部最小化。可以在有或没有坐标约束的情况下执行最小化。这些选项允许采用更准确和定制的方法来估计结合亲和力,而无需对接配体。
Rosetta 虚拟筛选 (RosettaVS) 筛选模式
RosettaVS 利用我们之前开发的运行模式GAligandDock 用于各种定制任务。在这项工作中,我们改进并引入了两种使用 RosettaGenFF-VS 进行快速配体对接和结合亲和力估计的运行模式。它们是 Virtual Screening eXpress (VSX) 和 Virtual Screening High Precision (VSH)。VSX 模式将蛋白质侧链视为刚性的,并在基因库大小为 50 的情况下执行五次迭代。该模式专为快速高效的筛选而设计。另一方面,VSH 模式允许在对接过程中使用灵活的口袋残基侧链,并在预先计算的能量网格上进行两阶段构象搜索。第一阶段将库仑相互作用权重增加三倍,并在基因库大小为 100 的情况下运行五次迭代。第二阶段使用库仑相互作用的默认权重,并在基因库大小为 100 的情况下运行五次迭代。在运行时,VSX 模式需要约 90 至 150 秒来筛选配体,而 VSH 模式的运行速度慢约六倍。尽管速度存在差异,但这两种模式都支持在单批次中对接多个配体。此功能通过读入多个配体的单个输入文件并输出多个配体的单个输出文件,显着减少了文件系统的负载。批处理大小的选择可以是任意利用本地计算集群的任意性。例如,在本研究中,我们使用VSX模式的批量大小为50,VSH使用5。在Rosettascripts58 XML格式的补充信息中提供了用于运行VSX和VSH的详细设置。
对ForceField和停靠协议的改进
我们对Rosetta General Forcefield(Rosetta General Forcefield)(Rosettagenff)和对接协议,以提高性能和准确性。新的原子类型和扭转类型被纳入Rosettagenff,以更好地建模三个和四元环。我们还解决了一些扭转潜力的问题,这些问题是由于某些扭转类型的不正确定义而产生的。此外,我们纠正了在配体对接中组氨酸的互变异状态期间发生的误差。此外,我们优化了小分子预处理脚本,以解决已知问题并增强其鲁棒性。This included accurately handling the atom typing of aromatic ring nitrogens, protonated nitrogens, and oxime oxygen atoms, as well as dealing with molecules with collinear structures and others.
Score function and virtual screening benchmarks
CASF2016基准
CASF2016的蛋白质结构通过使用Rosetta fastrelax协议59,60,61的坐标约束来预处理中。使用Ambertools 23.0S AnteChamber62处理CASF2016数据集中的配体数据集,以生成具有AM1BCC部分费用的Mol2文件。然后,使用Rosetta中的Mol2GenParams.py脚本将这些Mol2文件转换为Rosetta参数文件。由于该数据集没有采样过程,因此我们使用Rosettavs评估模式来评估Rosettagenff-Vs的得分函数性能。对于评分功率测试,通过坐标约束将配体和口袋侧链最小化。使用局部优化的结构,具有两个不同的熵模型,即简单的熵和默认熵,使用局部优化的结构估算了补充图2中报告的结合亲和力。结果表明,具有简单熵模型的Rosettagenff-Vs是基于物理学的基于结合亲和力预测的领先的得分函数。值得注意的是,Vinarf20使用了机器学习描述符。对于对接功率测试,使用坐标约束将小分子诱饵最小化。然后使用来自本地优化的结构的得分来计算对接成功。我们报道了图1D和补充图中的性能。3,7没有指定熵模型,因为Rosettagenff-Vs的对接功率不取决于熵模型的选择。如图1D和补充图3所示,Rosettagenff-Vs在配体对接精度方面取得了领先的性能。然后,我们在参考文献中进行了分析后进一步检查了结合漏斗。28.结合漏斗分析的目的是证明在最低能量最小值左右形成的漏斗状形状的质量。与对接精度不同,结合漏斗分析衡量了能量潜力驱动构象采样到最低能量最小值的效率。如补充图7所示,Rosettagenff在各种配体RMSD上表现出优质的结合漏斗,这表明与其他方法相比,更有效地搜索了最低能量的最低能量。对于筛选功率测试,允许诱饵在口袋内自由最小化,以反映现实世界虚拟筛选设置。该过程中预测的结合亲和力用于计算富集因子和成功率。结果(图1E,F和补充图4,5)表明,Rosettagenff-Vs在筛选功率测试中实现了最先进的性能。为了进一步研究筛选功率测试的改进性能,我们根据结合口袋内的排除体积(VOL)分析了三个子集的结合亲和力预测模型(VOL),掩埋了溶剂可访问区域的百分比结合后的配体(SAS)和结合口袋的疏水尺度(H尺度)。补充图8中的结果表明,与其他方法相比,几乎每个子集的结合亲和力方法在几乎每个子集上的表现均相同。此外,我们的方法在具有更大极(子集H1),较浅(子集S1)和较小(子集V1)口袋的目标蛋白上显示出显着改善,而其他方法通常表现不佳。在图1E和F中,我们报道了Rosettagenff-Vs-Simple作为Rosettagenff-Vs的最佳性能,以简单。尽管我们的主要比较是在这项工作中的其他基于物理的得分功能,为了提供对模型性能的更全面评估,但我们将模型与几种基于最新的深度学习功能进行了比较(请参阅补充数据1),其中Rosettagenff-Vs的票价很有优惠。
dud基准测试
有用的诱饵(dud)数据集的目录是从https://dud.docking下载的。org/https://dud.docking.org/。通过在提供的蛋白质PDB文件中以其相应的规范形式替换任何非典型氨基酸,然后使用Rosetta fastrelax方案放松,制备受体结构。通过将配体分子随机放置在每个受体中的结合袋中以指示结合口袋的位置,准备了Rosettavs的输入复合结构。小分子参数文件是根据DUD提供的Mol2文件转换的。例如,可以在补充信息中找到Rosetta脚本XML文件和Rosetta命令行。结果(图1b,c)表明,与其他虚拟筛选方法相比,Rosettavs中的VSX和VSH模式都取得了出色的性能。平均而言,与VSX模式相比,VSH模式的性能略有改善。我们检查了VSH模式提高性能并在补充图10中提供了两个示例的情况,强调了在配体docking中对灵活的侧级建模的重要性。
AI加速虚拟筛选协议
受体预处理
用于KLHDC2虚拟筛选,与SELK DEGRON肽(PDB:6DO3)复杂化的KLHDC2的晶体结构是从蛋白质数据库下载的。我们删除了所有溶剂分子,并保留了一个Degron结合的单体结构(链A和B)的副本。使用带有坐标约束的Rosetta Fastrelax方案将这种单体复合物放松。作为最后一步,我们用随机的小分子代替了弛豫结构中的C端Degron肽作为结合位点的指示。然后将这种修改的结构用作Rosettavs的输入。对于NAV1.7虚拟筛选,从蛋白质数据库下载了人类NAV1.7的冷冻结构 - Navab Channel Chimera(PDB:5EK0)26。去除所有溶剂分子后,我们保留了人NAV1.7- NAVAB通道Chimera VSD4(从M1493到P1617)的一个区域,该区域与配体GX -936直接相互作用并将其用作受体结构。使用带有坐标约束的Rosetta Fastrelax方案,使用配体GX-936将这种受体结构放松。然后将所得的放松结构用作Rosettavs的输入。重要的是要注意,GX-936仅用于指示Rosettavs的结合位点。
小分子预处理和库准备
rosettavs docking docking docking需要rosetta params params作为小的小参数。生成参数文件需要分子和小分子mol2格式。Enamine Real 2022-Q1库中包含约55亿个微笑,从Enamine(https://enamine.net/library-synthesis/real-compounds)下载。使用Dimorphite-DL64,在pH = 7.4处为质子化状态分配了微笑。然后将这些适当的质子化微笑字符串转换为使用OpenBabel65的Molecular 3D结构和MMFF94的部分电荷转换为Mol2文件。从Cartblanche22 Web服务器(https://cartblanche.docking.org)下载了大约41亿个现成的小分子mol2文件,其中包含约41亿个现成的小分子mol2文件。Rosetta中的mol2genparams.py脚本用于将Mol2文件转换为Rosetta参数文件,以用作Rosettavs中的输入。为了提高整个库的推论效率,我们为整个分子集合的指纹预先生成,并为它们准备对深度学习模型的投入做好准备。利用RDKIT66,我们生成了1024位Morgan指纹67,半径为2。
主动学习模型
我们使用基于简单的指纹的馈电神经网络实现了主动学习模型(FFN)用于训练粘合剂与非限制器的训练目标特异性分类模型。做出FFN的选择是为了降低替代模型的成本,模型体系结构和超参数搜索会经过未来的改进。该模型采用1024位指纹矢量作为输入,并输出一个表示分子为粘合剂的概率的单个值。FFN模型由两个密集连接的隐藏层组成,每个图层包含3000个节点。这些层之后是批准归一化,辍学率为0.5,以防止过度拟合。最后一层是线性的,然后是乙状结激活层,该层压缩了0到1之间的输出值,从而表示分子是粘合剂的概率。使用预算的指纹,使用单个CPU(Intel Xeon E5-2695 V3 @ 2.30 GHz),使用该模型的一百万分子的推断将平均使用,或使用RTX2080 GPU进行11 s。在整个主动学习过程中监视了模型性能,并且KLHDC2的最终模型的AUC为0.886,而NAV1.7目标的最终模型在独立的测试集上的AUC为0.927,这显示了最终的ML模型确实是每个目标的好二进制分类器。
OpenVS平台
我们开发了一个开源平台OpenVS,以简化整个AI-Accelerated虚拟筛选过程。该平台的一个关键功能是它可以使虚拟屏幕并行化并减少文件系统上的负载。这是通过通过Rosettavs协议中的多种配体对接功能将多个配体分解为单个虚拟屏幕作业的方法来实现的。通过在单个作业中对接N配体,输入和输出文件的数量有效地减少了N倍,从而大大降低了文件系统上的负载。此外,将多个配体批量组合在一起还减少了系统上的输入和输出(I/O)加载,因为并行运行需要更少的作业。该平台使用SLURM Workload Manager(https://slurm.schedmd.com)和GNU Parallel68用于虚拟屏幕的高平行化,相对于使用的CPU和节点的数量,虚拟筛选时间的线性缩放表现出。此外,该平台很容易适应支持其他工作调度程序,使其成为各种计算环境的多功能工具。
AI加速虚拟筛选的工作流程
在这项工作中,我们利用了主动学习技术指导探索广阔的化学空间。贪婪的策略用于选择每种迭代的新化合物,以增强培训数据集,而无需使用任何明确的不确定性信息来减少计算成本和推理时间。原则上,可以使用Monte Carlo(MC)Lotecout69通过在当前框架内使用激活的辍学层运行模型推理来获得不确定性估计。具体工作流如下所述。我们的第一步是创建一个专门的子集,该子集,称为含有〜4.936亿个分子的锌1570 3D药物样数据库,称为类似药物的基因库。该子集的创建涉及使用0.6 Tanimoto相似性的截止,从锌15 3D药物样数据库中聚类相似的分子。从每个群集中,选择了最小的分子并将其添加到库中,用作簇的质心。这一过程导致形成了类似药物的收科库,其中包括大约1300万个分子。创建类似药物的收科库的目的是确保模型在初始迭代期间暴露于广泛的化学空间。在第一次迭代中,从吸毒状的基督图书馆中分别随机选择了50万和100万个分子作为训练和测试数据集。我们使用Rosettav中的VSX模式将这些分子停靠在目标袋中。使用2和1024位的半径为所有分子作为深度学习模型的输入而产生的摩根指纹。我们为每次迭代选择了对应于测试集中最高N%的预测结合亲和力(DG)截止。此截止被用来分配粘合剂(