
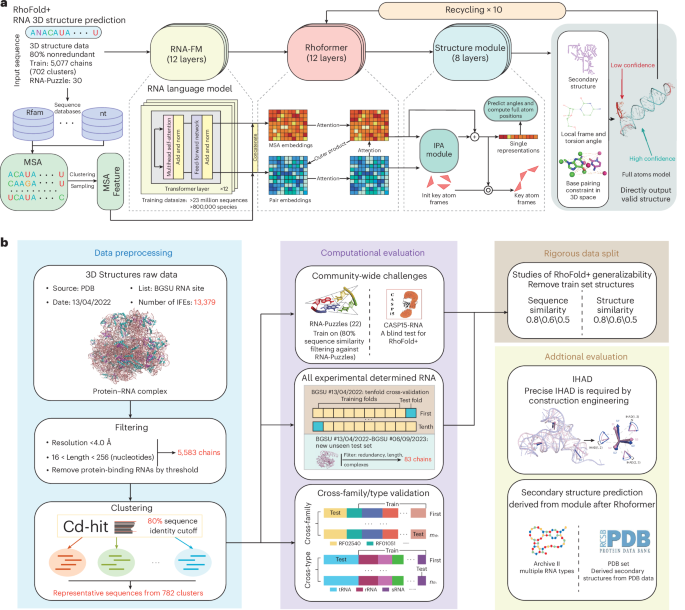
使用基于语言模型的深度学习方法准确预测 RNA 3D 结构
作者:Li, Yu
主要的
RNA分子在分子生物学的中心法则中占据着关键作用。RNA结构如何影响基因调控和功能一直是深入研究的课题1。专注于 RNA 靶向的研究表明,它可以成为药物开发的重要的可成药靶点2,3,4以及有用的合成生物学设计元素5。超过 85% 的人类基因组被转录,但只有 3% 编码蛋白质,这凸显了转录 RNA 的大部分具有未知的功能和结构。在许多情况下,获得高分辨率结构信息可以更好地预测性了解感兴趣的 RNA 分子4,6。
RNA 分子的构象灵活性使得其三维 (3D) 结构的实验确定具有挑战性。截至 2023 年 12 月,蛋白质数据库 (PDB) 约 214,000 个结构中,仅含 RNA 的结构仅占不到 1.0%,而含 RNA 的复合物仅占 2.1%(参考文献 1)。6,7)。尽管 X 射线晶体学、核磁共振波谱学和低温电子显微镜取得了进步,但这些低通量技术仍受到特殊要求的限制。计算方法已成为利用 RNA 序列数据进行 RNA 3D 结构预测的补充方法。这些方法分为两大类: 基于模板的建模,例如 ModeRNA8和RNA构建器9,受到有限模板库和从头预测方法(包括 FARFAR2)的限制(参考文献 1)。10), 3dRNA11和SimRNA12,由于大规模采样要求,预测能力更强,但计算量较大。
正交从头预测方法是利用深度学习,该方法已成功应用于各种生物问题。这些应用包括预测蛋白质 3D 结构13, RNA二级结构14,15并对其他方法生成的 RNA 结构进行评分16。以前的 RNA 3D 结构预测方法侧重于基于模板或基于能量的采样技术,这是由于可用 RNA 3D 结构数据的稀缺性。尽管数据稀缺,AlphaFold2 的成功(参考文献 2)13)用于蛋白质结构预测,促进了 RNA 3D 结构预测从头深度学习方法的发展。这些从头方法通常从单个输入序列开始,然后从中构建多个序列比对 (MSA),随后用于构建 3D 结构。
MSA 已被证明可以提供有助于蛋白质建模的额外信息,对于 RNA 来说可能同样如此。例如,DeepFoldRNA17 号和trRosettaRNA18利用变压器网络(例如 RNAformer)将构建的 MSA 和预测的二级结构转换为各种一维 (1D) 和二维 (2D) 距离、方向和扭转角。然后,利用这些预测的几何形状作为约束,使用能量最小化来预测 RNA 3D 结构,将采样和评分过程集成到其框架中。多种型号,包括 E2Efold-3D19和罗斯TTAFoldNA20,采用完全可微的端到端管道,使用构建的 MSA 和二级结构约束直接预测全原子 3D 模型。AlphaFold3(参考。21),AlphaFold2 的后继者(参考。22),还能够直接从输入序列预测 RNA 3D 结构,同时在预测过程中仍然依赖于其构建的 MSA。与其他方法相比,AlphaFold3(参考文献 1)21)采用基于扩散的过程来预测原始原子坐标,取代了对氨基酸特异性框架和侧链扭转角进行操作的 AlphaFold2 结构模块。虽然这些基于 MSA 的方法能够准确预测 RNA 3D 结构,但它们需要在大型序列数据库中进行广泛搜索,这可能非常耗时。相比之下,基于单序列的模型,包括 DRFold23,不使用 MSA,因此不需要在大型序列数据库中进行大量搜索。相反,DRFold23仅依赖于预测的二级结构来提供 3D 结构预测。与基于 MSA 的方法相比,此方法速度更快,但精度通常较低。下一代深度学习方法可能会更好地利用基于 MSA 的方法,从而提高速度和准确性。
在这里,我们提出了一种基于语言模型的深度学习方法 RhoFold+,用于准确、快速的从头 RNA 3D 结构预测。RhoFold+ 代表了对其前身 RhoFold 的完全自动化和可区分的改进19,利用 MSA 和其他功能的改进集成来提高性能。我们的主要重点是确定单链 RNA 的结构,单链 RNA 与其他分子的相互作用有限。应对这一挑战可以帮助我们更好地理解RNA生物学,并为解决更复杂的结构问题提供起点。
结果
用于 RNA 3D 结构预测的自动化端到端平台
RhoFold+ 的开发以 RNA 特异性知识和现有 RNA 3D 结构数据的局限性为指导。为了构建我们的训练数据集,我们使用 BGSU 代表性的 RNA 结构集(版本 2022-04-13)从 PDB 中整理了所有可用的 RNA 3D 结构24。我们专注于单链 RNA,并通过使用 Cd-hit 聚类序列来减少冗余25以 80% 的序列相似性阈值,从 5,583 个 RNA 链中产生 782 个独特的序列簇。然后,这些 RNA 序列通过我们的流程 RhoFold+ 进行处理。首先,使用我们的大型 RNA 语言模型 RNA-FM 转换序列,以提取进化和结构信息嵌入。同时,通过搜索广泛的序列数据库生成 MSA。然后将嵌入和 MSA 特征输入到我们的变压器网络 Rhoformer 中,并迭代细化十个周期。接下来,我们的结构模块采用了几何感知注意力机制和不变点注意力(IPA)模块来优化 RNA 主链中关键原子的局部框架坐标和扭转角。重建全原子坐标后应用结构约束,例如二级结构和碱基配对(图1)。1a以及详细讨论补充资料)。开发 RhoFold+ 后,我们在广泛的测试中对其性能进行了严格的基准测试和评估(图 1)。1b)。

, RhoFold+ 的架构,一种完全自动化且可微分的端到端方法,可从序列中进行 RNA 3D 结构预测。使用在 23,735,169 个未注释的 RNA 序列和多个深度学习模块(包括模拟 3D 位置的 IPA 模块)上预训练的 RNA 语言模型 (RNA-FM),RhoFold+ 可以生成有效且大致准确的感兴趣的 RNA 3D 结构,通常在 ~0.14 以内s(无 MSA 搜索)。init,初始化;规范,规范化。乙, RhoFold+ 的预处理步骤,用于从 PDB 数据库中提取所有可用的非冗余单链 RNA 3D 结构。IFE,集成功能元件。RhoFold+ 针对社区范围的挑战(包括 RNA-Puzzles 目标和 CASP15 天然 RNA 目标)以及所有可用的实验确定的 RNA 3D 结构进行了全面基准测试。RhoFold+ 还在交叉验证实验中表现出高精度,以及在跨家族和跨类型验证实验中对未见过的、新确定的 RNA 结构以及未见过的 RNA 家族和类型的通用性。数据分割评估表明,RhoFold+ 并未过度拟合其训练集。RhoFold+ 还能够预测对建筑工程有用的二级结构和参数。
在 RNA 难题上对 RhoFold+ 进行基准测试
我们对 RhoFold+ 和其他现有计算方法对之前举行的两项社区范围挑战:RNA-Puzzles 和 CASP15 进行了全面的回顾性比较。我们首先使用 RNA-Puzzles 的结果26,27,28,29,30竞赛,其中提交的作品是通过人类知识或计算方法生成和优化的。重要的是,这里 RhoFold+ 使用与测试的 RNA-Puzzles 目标相关的非重叠训练数据进行训练(方法)。我们进行预处理以获得 24 个单链 RNA 靶标并排除 RNA 复合物。这组 RNA 靶标包含两个谜题 (PZ):PZ34 和 PZ38,它们是在我们开发 RhoFold+ 后引入的(图 1)。2a和补充图。3),从而作为盲测。从官方服务器收集其他方法的预测后(http://www.rnapuzzles.org/),我们发现 RhoFold+ 在除 PZ24 之外的几乎所有目标上的性能均超过了所有其他方法(包括 FARFAR2/ARES)。值得注意的是,RhoFold+ 在一半以上的目标上比第二最佳方法的性能高出约 4 % r.m.s.d。在 17 个目标上,RhoFold+ 实现了 r.m.s.d。值<5-5,并且只有一个目标表现出 r.m.s.d。>10%(图 1)2a和补充表5)。总体而言,RhoFold+ 产生了平均 r.m.s.d。4.02%,比第二好的模型好 2.30%(FARFAR2:前 1%,6.32%)。使用模板建模 (TM) 分数进行评估31,RhoFold+ 平均达到 0.57(补充表5),高于其他表现最佳者的分数(0.41 和 0.44)。图 2:根据之前举办的全社区挑战对 RhoFold+ 进行基准测试。一个

RhoFold+ 和其他方法在 24 个非重叠、非冗余 RNA-Puzzles 目标上的性能散点图。每个点代表特定方法的预测模型。乙,RNA-谜题 7 和 38 的可视化。除了对齐的 RhoFold+ 预测之外,我们还展示了与每个目标最相似的训练结构,这表明 RhoFold+ 既不会过度拟合训练集,也不会简单地重现与目标最相似的结构。Seq-sim,序列相似性。c,RhoFold+ 预测的 TM 分数和 LDDT 与所有 RNA-Puzzles 目标的所有训练序列之间的最大序列相似性的回归图。每个点代表一个 RNA-Puzzles 目标。d, 不同方法的运行时间比较。e, 将 RhoFold+ 预测与我们的训练集中所有 RNA-Puzzles 目标的最佳单个模板进行比较。f, r.m.s.d 的回归图。跨所有 RNA-Puzzles 和 CASP15 靶标的原子级 pLDDT。克, 结构 GDT-TS 与所有 RNA-Puzzles 和 CASP15 靶标的 MSA 相似性的回归图。小时, CASP15 天然 RNA 靶标的详细性能比较。粉红色的柱子记录了详细的均方根误差 (r.m.s.d)。值和蓝色列记录了总和Z-GDT-TS 和 TM 分数。缺少官方报告的 CASP15 数据的条目被标记为 N/A;Yang-Sever 和 Chen 是 CASP15 注册团体。我, RhoFold+ 的平均性能与 CASP15 组和已发表的 CASP15 天然 RNA 靶标的平均报告性能的比较。j, 结构 GDT-TS 和 LDDT 与所有 CASP15 靶标序列长度的回归图。中心曲线在c,克和j表示拟合回归模型,而周围的两条曲线表示 95% 百分位数区间。k, RhoFold+ 预测与 AIchemy_RNA2 和 UltraFold 对来自 CASP15 的 R1116 靶标的比较。MSA-sim,MSA 配置文件相似性。我, 对于 R1156 目标,显示 RhoFold+ 潜在故障案例,涉及不正确的堆叠模式和方向。源数据。为了证明 RNA-Puzzles 上的有希望的结果不是由过度拟合产生的,我们研究了测试集和训练数据之间的序列相似性是否与 RhoFold+ 的性能呈显着正相关(通过 TM 分数和局部距离来衡量)差异测试 (LDDT),一种无叠加评分,用于评估模型中所有原子的局部距离差异
,33。以前在蛋白质结构预测中发现了这种相关性13,但在这里我们发现右2表示斜率是否显着非零的值,TM 得分为 0.23,LDDT 为 0.11(图 2)。2b、c),表明模型性能与我们的训练和测试集的相似性之间没有显着相关性。这些结果表明 RhoFold+ 可以概括地预测准确的 RNA 结构。一个代表性 RNA-Puzzles 靶点 PZ7(长 186 个核苷酸的 Varkud 卫星核酶 RNA)的案例研究证明了这一发现。这里,训练集中最相似的RNA的结构与PZ7的结构有很大不同(图1)。2b): r.m.s.d.这些结构之间的距离为 34.48°。作为另一个例子,PZ38 与我们训练集中的所有 RNA 相比表现出最高的序列相似性,为 53%,并且 r.m.s.d.序列最相似的RNA与PZ38的结构之间的差异为16.46±(图1)。2b)。这比 r.m.s.d 更大。PZ38 和 RhoFold+ 预测之间的差距为 8.92。
为了测试 RhoFold+ 对结构不相似(除了主要序列不相似)目标进行泛化的能力,我们试图确定 RhoFold+ 的预测是否可以超越训练集中的最佳单一模板(结构最相似的模型)给定的查询。为了研究这一点,我们将我们的预测和实验确定的结构之间的 TM 分数与所有 RNA-Puzzles 中最佳单一模板和实验确定的结构之间的 TM 分数进行了比较。对于大多数谜题,RhoFold+ 生成的预测具有更高的全局相似性,平均 TM 得分为 0.574,超过最佳单一模板 0.05(图 1)。2e和补充表13)。需要强调的是,对于蛋白质来说,超越最佳单一模板需要取得实质性进展。事实上,直到 CASP14 期间,计算方法才超越了最好的单一模板。尽管在传统序列相似性数据分割范式下,RhoFold+ 比其他方法生成的预测要准确得多,但我们通过从训练集中消除 3D 结构(其 TM 分数相对于任何目标超过指定阈值)进一步测试了 RhoFold+ 的适应性(补充如图。6和补充表6和10)。即使在这种更苛刻的条件下,RhoFold+ 仍然表现出有希望的性能(补充表10)。
在将计算模型应用于大规模的现实环境时,速度通常是重中之重。除了生成基本准确的折叠结果外,我们发现 RhoFold+ 速度很快,典型的 RNA-Puzzles 预测在约 0.14 秒内完成(图 1)。2d)。相比之下,其他方法,包括 SimRNA12, 远距离210和RNAComposer34,表现出明显更长的运行时间,可能是由于这些方法采用的大规模采样过程(图 1)。2d)。
在 CASP15 目标上对 RhoFold+ 进行基准测试
RNA-Puzzles 于十多年前首次发布26,我们接下来使用 RhoFold+ 来预测最近的 CASP15 中的 RNA 靶点(参考文献 1)。35,36)。我们重点关注 CASP15 的六个天然 RNA 靶标(图 1)。2小时和补充图。4)。不包括超出 RhoFold+ 预期应用范围的人工设计靶标:特别是,排除的靶标的特点是与我们的训练集缺乏同源性和差异,或者它们是 RNA-蛋白质复合物。我们遵循 CASP15 指南,其中规定参赛团队最多可以提交五个模型。利用不同的、随机采样的 MSA(方法),我们使用 RhoFold+ 为每个目标建模了五个候选结构,并仅考虑性能最高的预测(补充表6)。
几个排名靠前的CASP15小组和最近发表的RNA 3D结构预测工作17 号,18,20,21,23已包含在我们的基准测试中。特别是,根据是否使用人类专家知识和微调,CASP15 组被分为两类:“服务器”和“专家”。无论类别如何,许多 CASP15 小组都采用了基于自然目标的比较或统计学习的计算管道,从而使我们能够评估 RhoFold+ 的学习能力。我们的初步模型 AIchemy_RNA (RhoFold) 是“专家”类别的参与者。RhoFold+ 基于 RhoFold 构建,代表了一个完全自动化的端到端管道,更类似于“服务器”类别中的参与者。在这里,我们发现 RhoFold+ 在 CASP15 天然 RNA 靶标上的平均 r.m.s.d 优于 RhoFold。约 1%。此外,RhoFold+ 优于其他可预测所有 6 个天然 RNA 靶点的方法,包括排名第一的 AIchemy_RNA2、排名第二的 Chen 方法和其他计算方法,包括 DRfold23, 深折叠RNA17 号, 阿尔法折叠321和trRosettaRNA18(如图。2小时,我)。尽管 RhoFold+ 的性能略胜 AIchemy_RNA2 0.06%(平均 r.m.s.d.;图 1)。2i),AIchemy_RNA2 需要专业知识。此外,RhoFold+ 在几乎所有天然 RNA 靶点上都表现出与每种最佳方法相当的准确性(R1156 除外)(图 1)。2小时)。
遵循 CASP15 的评估方法36,我们还计算了Z-所有参与组的预测得分。CASP15 优先考虑 TM 分数和全局距离测试总分(GDT-TS),它评估整体结构相似性和局部对齐,使我们根据累积的值来评估这些模型Z- 这些指标的分数(图 1)2小时)。在六个天然 RNA 目标以及所有 CASP15 参与者对这些特定目标进行排名的子集中,RhoFold (AIchemy_RNA) 排名第四,而 RhoFold+ 的性能与 AIchemy_RNA2 相当(在Z-score)并超越了其他方法。在对特定目标的性能进行详细分析时,我们发现,对于目标 R1108,RhoFold+ 取得了最佳成绩Z-分数和rmsd。有趣的是,RhoFold+ 也获得了最佳成绩Z-R1116 的得分,尽管 r.m.s.d.比 UltraFold 高约 1%(其他方法产生的预测精度明显较低,均方根偏差 >10%)。经过进一步调查,我们发现,虽然 UltraFold 通过生成准确的局部预测在该指标上优于 RhoFold+,但预测的全局结构不太准确,TM 得分为 0.497,GDT-TS 得分 <0.4 就证明了这一点。相比之下,RhoFold+ 不准确地预测螺旋角,导致 r.m.s.d.8.92%,但其正确预测的拓扑导致 TM 得分 >0.55。对于该目标,AIchemy_RNA2 错误地预测了茎堆积和 RNA 拓扑结构,导致较高的 r.m.s.d。17.26 分,TM 分数约为 0.49。值得注意的是,R1116 的 RhoFold+ 预测并不是由过度拟合引起的,如 R1116 相对于训练集的较低的最大结构相似性(TM 得分)和最大序列相似性所示(图 1)。2k和补充表6)。
我们还研究了 RhoFold+ 可能会降低性能的目标,发现较高的 MSA 质量与更好的性能相关。虽然 RhoFold+ 准确地预测了局部结构拓扑,但它在对齐螺旋方面遇到了困难,特别是在连接处。这种差异可能是由于 RNA 连接的动态和灵活性质造成的,RNA 连接通常采用多种构象37,38,39,这使得全自动模型难以准确表示(图 1)。2k,升以及详细讨论补充资料)。
影响预测精度的因素
基于上述发现,我们进行了一项更全面的研究,涉及所有 CASP15 天然 RNA 和 RNA-Puzzles 靶标。我们观察到,RhoFold+ 的预测准确性对查询的 MSA 配置文件相似性很敏感(补充资料)针对训练集(图 2)2克)和RNA结构的复杂性(查询长度;图1)2j)。此外,预测的 LDDT (pLDDT) 分数被发现与 RhoFold+ 的置信度相关,为识别预测精度较低的区域提供了有用的指标,特别是在更复杂或不太同源的查询中(图 1)。2f以及详细的讨论和分析补充资料)。
对所有确定的 RNA 3D 结构进行 RhoFold+ 基准测试
在使用 RNA-Puzzles 和 CASP15 对 RhoFold+ 进行基准测试后,我们接下来使用所有实验确定的 RNA 结构(由 BGSU 代表性 RNA 结构集定义)(经过预处理以消除冗余)更详细地评估 RhoFold+。为了进一步研究 RhoFold+ 的性能,我们通过迭代屏蔽 80 个序列簇进行验证并留下 702 个序列簇进行训练,进行了十倍交叉验证。我们发现,无论训练和测试数据如何分割,RhoFold+ 在所有 RNA 结构中的性能都很稳健,并且在所有折叠中都相当一致(图 1)。3a~c)。TM 分数的轻微变化可能是由具有挑战性的目标引起的,例如与 PZ24 类似的 Fold2 和 Fold7 中的假结情况(图 1)。3c,e),我们预计,如果提供二级结构约束,RhoFold+ 对此类目标的预测可以得到改善。此外,在我们的交叉验证测试中,RhoFold+ 的准确预测不仅仅是因为模仿了最序列相似的训练数据(图 1)。3b、d、e)。r.m.s.d 的绘图相对于序列长度表明 r.m.s.d.值大部分分布在 10 以内,与序列长度无关(图 1)。3a)。具有 r.m.s.d 的离群值对于长度超过 200 nt 的序列,更有可能出现 >20 nt 的情况,我们期望通过对长 RNA 进行更多调整来进一步改进(详细讨论见补充资料)。

, r.m.s.d. 绘图所有交叉验证实验的序列长度值。每个点代表一个 RNA 结构,并根据交叉验证折叠进行着色。乙,针对所有训练数据的最大序列相似性,对每个预测的 TM 分数(蓝色)和 LDDT(粉色)进行回归分析。每个点代表一个 RNA 结构。c,每次折叠的平均 TM 分数和 LDDT。d,两个代表性核糖开关结构 6UES 和 3UD4 以及假结 1DDY(粉色)的可视化,以及相应的 RhoFold+ 预测(石板)和具有最高序列相似性的训练 RNA 结构(青色)。在一个—d,显示了使用所有实验确定的 RNA 结构对 RhoFold+ 进行的十倍交叉验证。e,新确定的 RNA 结构 7QR3 的可视化,7QR3 是一种丁型肝炎病毒 (HDV) 样核酶,其与训练集的结构相似性较低,但 RhoFold+(石板)可以准确预测其结构(粉红色)。最相似的结构 7DLZ 以青色显示。f, 平均 r.m.s.d 的比较RhoFold+ 和其他方法在新 PDB 集(一组 76 个新确定的单独 RNA 结构)上生成的值。克, 预测 r.m.s.d 的回归图。针对 RhoFold+ 和其他基线方法的训练集的最大序列相似性的值。小时, RhoFold+ 预测 TM 分数/LDDT 与训练集的最大 MSA 配置文件相似度之间相关性的回归图。中心曲线在乙和小时表示拟合回归模型,而周围的两条曲线表示 95% 百分位数区间。我, 通过 LDDT 和 TM 评分衡量的 RhoFold+ 交叉类型验证性能概述。用于验证的类型中的所有结构在模型训练期间都被屏蔽。sRNA,小RNA。j, RhoFold+ r.m.s.d. 的小提琴图。跨家庭验证中的价值观。在这里,待测试家族中的所有结构在模型训练过程中都被屏蔽,RhoFold+ 准确预测了大多数看不见的家族的 RNA 结构。每个家族中的序列数显示在括号中。
作为对 RhoFold+ 功能的进一步评估,我们考虑了该模型在训练数据集编译后发布的新确定的 RNA 单链结构上的性能。这种方法充当额外的盲测,类似于 CASP15 竞赛。我们将 FARFAR2 和最近的深度学习方法进行了比较17 号,18,21,23,所有这些都有可用的推理代码和/或服务器,其中一些还参与了 CASP15 (方法)。RhoFold+ 的性能优于所有基准模型,达到了以 r.m.s.d 衡量的最高平均准确度。RhoFold+ 产生平均 r.m.s.d.7.74%,分别比排名第二的 DeepRNAFold 和排名最低的 FARFAR2 好约 0.8% 和 10.5%。值得注意的是,平均而言,RhoFold+ 的性能也比 AlphaFold3 和 RoseTTAFold2NA 分别高出约 2.2% 和 1.8%(图 1)。3f以及详细讨论补充资料)。这些结果与我们之前在 CASP15 基准测试中观察到的性能一致,表明 RhoFold+ 可以准确地推广到我们的训练集中未见的新确定的结构。此外,这些结果表明,旨在预测生物分子复合物的 AlphaFold3 和 RoseTTAFold2NA 在应用于单个 RNA 分子时,表现不如 RhoFold+。进一步检查与我们的训练集的序列和结构相似性表明,即使序列相似性低于 0.5,RhoFold+ 仍保持强劲的性能(图 1)。3克),并且TM得分很大程度上受到MSA轮廓相似性的影响,而局部准确度(LDDT)仍然很高且稳健(图2)。3小时)。此外,尽管 RhoFold+ 与最接近的训练模板 7DLZ 的相似度较低(TM 得分为 0.40,r.m.s.d. 为 16.45%),但 RhoFold+ 仍表现出很强的泛化性,可准确折叠结构(例如 7QR3);图 13e)。
RhoFold+ 推广到看不见的 RNA 类型和家族
在证明 RhoFold+ 可以推广到预测具有不同序列相似性、结构相似性和发布日期的 RNA 结构后,我们接下来研究了 RhoFold+ 处理由专家知识定义的不同 RNA 类型和家族的能力。特别是,RNA类型和家庭(例如在RFAM中策划的家庭)40通常是根据功能,结构和共同进化信息在内的因素进行分类的。解决对不同RNA类型和家庭的概括的挑战可能对诸如Rhofold+之类的深度学习方法的要求更高,因为这项任务需要更大的领域转移。
我们通过对其他RNA类型的子集进行训练,在对其他RNA类型的子集上训练该模型,从而基准了Rhofold+的跨型性能。Rhofold+在RNA类型中表现出鲁棒性。尽管在内含子和核糖开关中挣扎,但它在转移RNA(TRNA)和微RNA(miRNA)类型上表现良好,达到了TM得分高达0.73(图。3i)。与Farfar2相比,Rhofold+在所有RNA类型中都优于它,尤其是在TRNA和核糖体RNA(RRNA)中,核糖开关的边缘较小(详细讨论补充信息)。对于跨家族测试,Rhofold+达到了平均R.M.S.D.6.69â(图。3j),但与复杂家庭(例如I组内含子(RF00028))挣扎。这种困难与在跨型测试中观察到的挑战一致,例如内含子和CRISPR RNA元素等复杂的RNA类型(RF01344)。这些元素与各种蛋白质和酶相互作用,仅专注于RNA结构而不考虑这些相互作用可能会限制预测准确性(详细讨论补充信息)。总体而言,这些测试证明了Rhofold+跨越看不见的RNA类型和家庭概括的能力,尽管对于具有有限的可用数据的复杂结构和数据集仍然存在挑战。
Rhofold+预测二级结构和子结构
Rhofold+可以准确预测RNA 3D结构,但是实验确定的RNA结构和类型数量有限,因此很难理解所有可能的RNA折叠的空间。对于复杂且大的RNA类型,包括内部核糖体进入位点,内含子,合成RNA和长期非编码RNA,尤其如此。但是,RNA二级结构可以在实验中更容易确定,而准确的二级结构预测可以补充3D结构的预测,从而为RNA折叠和功能提供了宝贵的见解。因此,我们对Rhofold+进行了调整以预测二级结构。由于Rhofold+旨在预测RNA 3D结构,因此我们合并了一个后处理模块,该模块利用了Rhofold+的Rhoformer检索到的特征来预测二级结构(因为Rhoformer的特征显示了注意力图,与接触式地图高度排列;补充图;补充图;补充图;如图。8和补充表14)。该模块考虑了与执行3D重建的模块相同的结构信息,但在对预测二级结构的不同几何和生物学约束下运行。
我们在新确定的PDB结构(新的PDB集)和Archiveii数据集上基准了Rhofold+的性能41,其中包括用于不同RNA的二级结构信息。在新的PDB套装中,Rhofold+优于表现的Ufold41平均F1分数为0.035(图。4a),即使对所有可用数据(PDB和BPRNA-1M,一个超过100,000个注释的RNA二级结构)进行了培训(PDB和BPRNA-1M)。在包含2,975个RNA样品的档案数据集上,Rhofold+也优于其他二级结构预测方法(图。4b),特别是在较大的RNA类型上(图。4c)。例如,它在登革热病毒转录组中的结构化结构域中达到0.60的F1得分(补充表19),与突变分析的结果(环图)对齐42,43。同样,Rhofold+的强烈性能也不源于模仿训练数据,因为即使序列相似性下降到50%以下时,它的F1得分也约为0.7(图。4e),并在CASP15目标R1117上获得了1.0的完美F1得分(图。4f)。这些结果表明,Rhofold+不仅在预测3D结构方面表现出色,而且还产生了丰富的,有意义的表示,可以实现最先进的二级结构预测。

,F1分数比较与PDB集中的UFOLD多种配置。在这里,在BPRNA上训练的UFLOLD版本也作为基线提出,以评估F1分数的改进。乙,在ArchiveII数据集上的各种方法的F1分数分布。平均分数在图的顶部指示。c,rhofold+和ufold在ArchiveII数据集上的F1分数比较。每个点代表RNA结构,并根据其RNA类型进行着色。SRP,信号识别粒子RNA;TMRNA,转移给我的RNA。d,在新PDB集合中,Rhofold+与Ufold和ufold和spot-RNA的F1分数比较。e,在新PDB集合中,Rhofold+与UFOLD和SPOT-RNA的F1得分比较与RNA结构的序列相似性。f,可视化CASP15 RNA靶标,其中Rhofold+预测了包括伪KNOTS在内的正确二级结构。克,可视化交换的二聚体,四氢叶酸(THF)核酶,3SUH,为此,Rhofold+预测(紫色)类似于生物学上有意义的结构(橙色),而不是在PDB中发现的晶体图形(橙色)。小时,可视化显示IHAD的定义,这是源自Rhofold+预测的IHA与实验确定的结构之间的差异。我,IHAD和R.M.S.D.之间的回归分析Rhofold+预测。每个点代表一个RNA。j,源自Rhofold+预测与实验结构的IHA之间的比较。每个点代表一个角度实例,并根据R.M.S.D进行着色。在包含角度+预测的角度的实验结构之间。k,IHAD对实验确定的IHA值的图。着色与j。中央曲线e,j和k代表拟合回归模型,而周围的两个曲线表示95%的间隔。源数据。
44和Ufold41在所有子结构中,多轨和外部循环的最显着改善,而内部环和伪诺斯在跨方法中显示出相似的性能(图。4天)。这些结果强调了Rhofold+在预测RNA二级结构并增强我们对RNA功能的理解方面的潜在能力。
纠正工件和IHA预测
正如Rhofold+准确地预测了二级和三级水平上的RNA结构,我们询问是否可以利用Rhofold+进行实验工作。在此方面,我们研究了两种用餐案例+:(1)用于校正实验结构伪影,(2)用于指导RNA构建工程。
X射线晶体学广泛用于解析RNA 3D结构,但可以引入诸如域交换二聚体之类的伪影45,潜在的误导机器学习模型不会很好地概括。在一种情况下,3SUH的Rhofold+预测最初产生了高度的R.M.S.D.与PDB结构相比,为10.11â。然而,进一步的分析表明,晶体结构涉及域交换的二聚体。当将Rhofold+预测与推断的单体结构进行比较时,R.M.S.D.提高到5.71ââ,表明Rhofold+准确地预测了与生物学相关的结构(图。4克)。ZTP Riboswitch也观察到了类似的发现46(补充图。9),表明Rhofold+可以有效纠正此类实验工件。
在将实验数据与RNA 3D模型进行比较时,其他几何指标(例如间旋转角度(IHAS))可以提供超出标准全局比对度量(例如R.M.S.D.,LDDT和TM得分)的见解。可以使用实验方法估算的IHA,可用于验证预测模型和指导RNA纳米结构设计。我们引入了IHA差异(IHAD)作为度量标准,以基于Rhofold+的预测(图。4小时和补充信息),发现IHAD可以揭示R.M.S.D.未捕获的STEM方向的差异。单独(图。4i)。我们的分析表明,Rhofold+通常准确地预测了茎方向(图。4J,k),尽管在0°或180°附近的IHAS的性能下降,但可能是由于大而复杂的结构中平行茎不足所致(图。4k和详细讨论补充信息)。我们通过预测来自四环胺I组I内含子的RNA构建体的值(例如FMN核糖开关和P4 p6域),进一步证明了IHA的实际应用(补充图。9)。
消融研究和产生多个预测
鉴于Rhofold+的高精度和速度,我们最终进行了消融研究,以了解哪些组成部分和信息对Rhofold+预测很重要。我们研究的结构组件包括四个不同的模块(图。5a和方法)。对138个PDB靶标(2022年4月至2023年12月之间收集)进行了消融研究,序列相似性低于我们的训练集,长度为16至300 nt的长度(烧蚀集)。通过删除每个Rhofold+组件,我们观察到所有这些都有助于提高性能,而MSA模块是最关键的,其次是RNA-FM语言模型(图。5a)。没有MSA模块的Alphafold2的RNA修饰版本比Rhofold+更差(图。5a)。值得注意的是,去除RNA-FM导致不同序列的性能下降急剧下降(图。5b),RNA-FM模块似乎可以补偿MSA模块的损失,从而保持了更高的TM分数(图。5c)。此外,删除回收模块对更长序列的预测最显着影响,这可能是由于其在有效加深模型中的作用(补充图。7和详细讨论补充信息)。

,Rhofold+没有(w/o)相应模块的Rhofold+的消融研究,其性能由R.M.S.D测量。乙,针对序列相似性倒数的预测准确性(由R.M.S.D.衡量)的回归分析。c,对于RNA-FM模块的消融研究,对MSA深度的TM得分进行了回归分析。请注意x轴是日志缩放的。d,针对MSA深度的预测准确性(通过TM分数衡量)的图。e,跨不同MSA深度的Rhofold+对Rhofold(由R.M.D.的测量)的改进图。f,在不同的MSA轮廓相似性上,Rhofold+对Rhofold(由R.M.S.D)的改进图的图。中央曲线e和f代表拟合回归模型,而周围的两个曲线表示95%的间隔。克,可视化CASP15目标,其中Rhofold+产生R.M.S.D.使用MSA采样的TOP5预测,但使用了12.51â,但提高了8.92â。小时,可视化新确定的RNA结构,其中R.M.S.D.使用MSA采样的TOP5预测,Rhofold+的Rhofold+提高了7.92â。
这些发现与我们对CASP15的天然RNA靶标和RNA-Puzzle的结果一致,其中MSA质量显着影响预测。我们还探讨了提取的MSA中序列的数量如何影响精度。尽管由于训练限制,Rhofold+限制为256个MSA,但该限制并没有损害其有效性。Rhofold+的关键增强功能是它通过从固定数量的MSA中取样或聚类来生成多个预测的能力,从而可以进行更广泛的预测选择和改进的结果。在RNA puzzles上的性能显示出与降低的MSA计数的反相关性,当MSA数超过100时,其显着改善(图。5天),表明较大的MSA池增强了模型优化(详细讨论补充信息)。随着MSA采样的扩展,最低的R.M.S.D.与Rhofold相比,Rhofold+ Top5的预测显着降低,与MSA深度的增加呈正相关,并产生高达10的改善(图。5e)。当查询和训练序列之间的MSA曲线相似性很高时,这种改善就会更加明显,而当相似性已经很强时,就会导致较小的增长(图。5f)。总体而言,其他MSA采样对于高性能至关重要,如Casp15目标R1116和PDB 7VPX_L所证明的那样(图。5G,h)。
讨论
在这项研究中,我们开发了一种基于端到端语言模型的深度学习方法Rhofold+,以预测序列的RNA 3D结构。Rhofold+是一种完全自动化和可区分的模型,它集成了在约2370万个RNA序列上预处理的RNA语言模型,而无需结构信息泄漏和多种策略来增强稀缺训练数据。Rhofold+优于其他RNA结构预测方法,基于对CASP15天然RNA靶标的深度学习,并实现了低于4的平均R.M.M.S.D.用于非重叠和非冗余的RNA-Puzzles结构。由于Rhofold+不需要任何时间耗时和计算密集的抽样过程,因此Rhofold+也是快速有效的,并且它也不依赖于专家知识,而专家知识已用于到目前为止最高表现的RNA结构预测。Rhofold+能够从不同的训练数据集中概括,并准确预测可用的RNA 3D结构和新确定的结构,这是由Rhofold+在交叉验证过程中强大的鲁棒性强调的观察结果。此外,Rhofold+可以在跨家族和跨型验证期间准确预测看不见的RNA结构。尽管Rhofold+旨在预测3D结构,但它也可以准确预测RNA二级结构。应用Rhofold+预测IHAS的一项由低温电子显微镜和基于NMR的构造工程设计启发的任务,这表明其潜力有可能加速实验确定更多RNA结构的过程。
尽管Rhofold+显示出令人鼓舞的性能,但它与其他深度学习方法共享了RNA结构预测的局限性。首先,我们对RNA结构多样性的了解是有限的,这使得由于它们的动态性质和与其他分子的相互作用,预测同一RNA分子的不同构象。例如,RNA连接可以采用多种构象,并且可以更好地表示为动态合奏37,38,39。其次,由于数据不足,可以预测大而复杂的RNA结构,尤其是具有多个螺旋或伪数的大型RNA结构,这仍然很困难,尤其是对于超过500个核苷酸的序列而言。第三,涉及配体或蛋白质的RNA复合物提出了其他挑战,因为当前方法通常无法充分解释这些相互作用,从而降低了准确性。而诸如alphafold3之类的方法(参考。21)和Rosettafoldna20可以预测RNA复合物,其准确性仍然有限,并且在单链RNA上的性能不如Rhofold+。第四,Rhofold+和类似模型在从特定环境条件的数据集上进行了训练,这可能无法很好地推广到RNA分子在体内遇到的各种和动态溶液条件。这些条件包括不同浓度的离子,例如镁和钾,以及配体的存在,这些配体在RNA折叠和稳定性中起着至关重要的作用。
依赖MSA的方法受这些比对的可用性的限制,这使得缺乏相应MSA的人工设计或孤儿RNA难以准确的预测。尽管RNA-FM有助于减轻这种依赖性,但仍然存在挑战。Rhofold+和类似的深度学习模型虽然准确,但由于对RNA结构多样性的了解有限,在预测大型和复杂结构方面的困难以及对MSA的依赖。为了减轻这些障碍,整合探测方法以定义二级结构,结合分子动力学和能量功能技术以及改善MSA提取过程可能会提高Rhofold+的准确性。此外,解决RNA蛋白和RNA配体相互作用仍然至关重要,并且将Rhofold+与蛋白质结构预测工具(如Rosettafoldna或Alphafold3)相结合可以提高其在这些区域的能力。方法
Rhofold+平台
MSA功能生成
我们使用了地狱构建的MSA
47和RMSA(https://github.com/pylelab/rmsa)捕获序列的共同进化信息作为附加输入。使用地狱,可以找到具有保守二级结构的同源序列,而另一方面,RMSA采用基于RNA序列数据库的迭代搜索策略。我们利用了核酸序列数据库RFAM和rnacentral48。在Alphafold2中,使用了类似的方法与不同的对齐工具和序列数据库使用。鉴于需要产生多种模型和硬件存储器施加的约束,我们将完全提取的MSA减少到训练阶段最多256个序列。随后,在推论阶段,通过聚类随机选择或选择256个MSA,然后喂入Rhofold+。我们通过验证的RNA语言模型从保守的二级结构或序列嵌入实施聚类。因此,可以将不同的采样和聚类结果用于多个预测,如top5,top10等。默认情况下,选择了前256个MSA作为预测标准结构的输入功能,我们将其称为标准Rhofold+。Rhofold+(顶部K)指从中选择的最佳模型K使用不同采样的MSA生成的不同模型。
RNA-FM语言模型
RNA-FM的概述
我们的基础模型提供了从独立序列信息推断出的有意义的表示。这些表示可能会改善各种下游任务的性能,尤其是对于那些没有足够注释数据的人。受最近研究的启发49,50,我们利用一般的变压器体系结构。特别是,我们的框架建立在BERT中提出的双向变压器语言模型(来自Transformers的双向编码器表示)中51,然后是无监督的培训计划。我们命名了我们的框架RNA-FMâ,因为它代表了未来RNA相关研究的基础模型(补充图。2)。下面,我们详细介绍了如何构建大型非编码RNA(NCRNA)数据集,然后进行模型和培训细节。
大规模预处理数据集
从rnacentral收集了预处理阶段中使用的大规模数据集48,迄今为止最大的NCRNA数据集。该数据集是NCRNA序列的综合集合,代表来自各种生物的所有NCRNA类型。它结合了跨47个不同数据库的NCRNA序列,总共约有2700万个RNA序列(补充表24)。
我们通过用u替换所有实例来预处理所有NCRNA序列,因为它们都与腺嘌呤互补,并且在结构上相似(代表DNA中的胸腺氨酸,而同时u As是RNA中的uracil)。这导致了一个涉及四种主要基础类型的数据集(总共16种计算类型的组合类型:y,y,kâ,,nâ和)。此外,为了最大程度地减少冗余而不损害数据集的大小(也就是说,为了保留尽可能多的序列),我们使用CD-HIT-EST删除了重复序列,CD-HIT-EST将其设置为100%的相似性阈值。在上述预处理步骤之后,获得了由超过2370万个NCRNA序列组成的最终大规模数据集。我们命名了最终数据集rnacentral100â,我们使用此数据集以自我监督的方式训练我们的RNA基础模型(请参阅补充信息了解更多详情)。
RNA-FM培训细节
我们的RNA-FM框架包括12个变压器编码器块,灵感来自Bert49,51。每个块包括一个640个隐藏尺寸的进料前层和一个具有20个头部的多头自我发言层,以及层归一化和剩余连接,分别应用了前后的损耗。对于长度的RNA序列L,RNA-FM将原始的顺序令牌作为输入,通过嵌入层将每个核苷酸映射到640维矢量中,形成一个L640嵌入矩阵。该矩阵穿过每个编码器块,在整个过程中保持其大小,然后是软磁层,以预测相应的令牌,包括16个核苷酸和四个特定功能标识符。其他模型详细信息在补充信息。
在训练期间,我们采用了类似于伯特的自制培训51,用特殊的面具令牌随机代替15%的核苷酸令牌。如果我选择了Token,将其替换为(1)(1)80%的时间,(2)随机令牌的时间为10%,(3)剩下的10%的时间不变。我们使用蒙版语言建模(MLM)训练了模型51,通过跨透镜损失预测原始的蒙版令牌。该培训策略作为目标函数提出如下:
$$ {{\ Mathcal {l}}} _ {\ Mathrm {mlm}}} = {{{\ Mathbb {e}}}}} _ {X \ sim {\ sim {\ Mathcal {\ Mathcal {x}}}}}}}}}}} _ {{X} _ {{\ Mathcal {m}}}} \ sim X} \ sum _ {i \ in {\ Mathcal {m}}}}}}}}} - \ log p({x} _ {x} _ {i} | {i} |{x} _ {/{\ Mathcal {m}}})。$$
(1)
一组索引\({\ Mathcal {M}}} \)从每个输入序列随机采样x,覆盖序列的15%,并用面具令牌代替相应的令牌。对于每个蒙版令牌,给定蒙版序列(\({x} _ {/{\ Mathcal {m}}}} \))作为上下文,目的函数最大程度地减少了真核苷酸的负模样x我。这种方法捕获了序列的掩盖和未掩盖部分之间的依赖性,从而准确地预测了掩盖位置。在方程式中使用目标函数的培训(1)允许RNA-FM有效地建模每个顺序令牌的表示。我们使用0.0001的基本速率,0.01的重量衰减和10,000个热身步骤训练了1个月的八个80 GB A100图形处理单元(GPU)的RNA-FM 1个月。为了优化内存使用情况和批处理大小,我们将最大输入序列长度设置为1,024,从而加速了训练过程。
有效开发自我验证数据集
尽管我们的RNA-FM可以减轻数据稀缺性问题,但与蛋白质相比,RNA的结构数据仍然少。结果,我们从rnastralign和bprNA-1M数据库中收集了一个非冗余,自我验证数据集,并具有地面真理二级结构。我们通过去除超过256或不到16个核苷酸的序列来过滤该数据集,从而导致数据集为27,732个序列。最初仅使用PDB数据对Rhofold+进行训练,然后通过推断伪结构标签来生成自鉴定数据集。我们通过对PDB数据的25%和75%的蒸馏数据进行取样,以进一步改进来重新训练该模型。在训练过程中,我们掩盖了PLDDT分数<0.7的伪标签残基,并均匀地采样了MSA,以增强蒸馏数据集。
结构预测模块
Rhofold+的结构模块旨在基于序列和由Rhoformer提取的配对表示预测RNA的3D结构。Alphafold2的结构模块直接预测了主链框架的旋转和翻译矩阵,因为这些是蛋白质折叠中最具影响力的因素。但是,RNA折叠主要由核苷酸碱基配对驱动。由于其不规则的结构模式,直接预测基础框架(\({\ Mathrm {C}}} {1}^{{{\ prime}},{\ Mathrm {N1/N9}},{\ MathRM {C2/C4}}} \))在核苷酸上定义的定义可能会在我们的实验中提出收敛问题(补充表1)。为了有效地重建RNA全原子坐标,我们使用了框架(\({\ Mathrm {C {4}}}}^{{{\ prime}},{\ Mathrm {C {1}}}}}}^{{\ prime}},{\ Mathrm {N1/N9}}}}}}}}} \))和四个扭转角度±, -,γ和 -解决这个问题。补充表1提供扭转角度和相应刚性组的定义。3D位置是使用IPA建模的,即几何学意识到的注意操作。根据Rhoformer的输出功能和配对的表现,IPA操作预测了每个帧的旋转和翻译矩阵。此外,使用回收策略对预测的结构进行了迭代的完善,其中rhoformer从上一个迭代中获得了预测。当PLDDT是IPA产生的输出之一,该输出量衡量了预测的3D结构的质量。通过重建的全原子坐标,可以直接在3D空间中执行生物学约束,例如基础配对,以优化结构模块并生成生物学上有效的结构预测。
使用Rhoformer的功能处理
与AlphaFold2中引入的EvoFormer一样,我们的主要模块Rhoformer由一系列带有封闭式自发层的变压器模块组成,这些模块被用来学习进化信息,并同时更新了成对序列嵌入和MSA表示。将包含两个线性层的过渡块添加到结果对和MSA表示中,以使嵌入维度增加四倍,从而增加模型的容量。最后,将四个自我发项障碍块堆叠在犀牛上,以完善对和MSA表示。然后将这些表示形式送入结构模块中,以在三维空间中获得预测的完整原子坐标,如以下各节所述。
结构预测损失
损耗函数定义为1D,2D和3D水平。这些级别中的每一个将在下面进行详细讨论。我们首先雇用了传销L传销为了改善从1D级别从MSA中提取共同进化信息,而无需添加策划的相关特征。在我们的实验中,将5%的核苷酸被随机掩盖,并利用线性投影层重建它们。
在2D级别,距离损失L迪斯和二级结构损失LSS应用于监督Rhofold+以学习每个残基之间的成对位置相关性。特别是,将三个进料层用于距离预测,以预测P,C4和N原子之间的成对距离。距离分为40个垃圾箱,第一个和最后一个垃圾箱分别表示<2â2â和>38ââ,而2â和38â之间的距离均匀地分为36个垃圾箱。另外,使用跨透明镜损失来确定距离预测是否属于正确的bin。对于二级结构预测损失LSS,在成对特征的顶部利用了一个前馈层,以预测二级结构。二级结构C是一个L -L二进制矩阵,哪里L表示序列长度,并且C我,j= 0或1表示我th 和j碱基对的残留物。在3D级别上,梯度源自主帧对齐点误差(FAPE)损失,表示为
LFAPE,二级结构约束损失和违反冲突损失L冲突。Alphafold2的FAPE损失比较了一组预测的原子坐标,并在一组预测的本地框架下与相应的地面真理原子坐标和地面真相局部框架进行了比较。损失与刚性动作无关。当预测的结构通过任意旋转和翻译与实际结构不同时,损失保持恒定。
二级结构约束损失,LSS3D,将次级结构信息直接编码为3D预测。为了统一3D空间中不同类型的基本配对约束的计算,我们在基本的局部坐标系中引入了四个固定的伪原子(T1,T2,T3和T4)52(补充图。1)。LSS3D旨在限制两个碱基对核酶中的伪原子,以满足碱基配对特性(基础碱基相互作用)。对于两个残留物米和n,我们计算了固定点的成对距离:\({{\ Mathbb {d}}}}}^{m,n} = \ {{d} _ {i,\,j}^{m,n} | i,j \ in \ in \ in \ {1,2,3,4 \} \} \), 在哪里米和n表示两个RNA残基,我和j是四个原子的索引。我们定义了LSS3D如下:$$ {l} _ {\ mathrm {ss3d}} = \ mathop {\ sum} \ limits _ {\ begin {arnay} {array} {c} m = 1 \ \ \ n = 1
_ {{\ rm {nbpairs}}}}}} \,\ max \ left({\ hat {d}} _ {i,\,\,j}^{m,n} - \ tau - {{d}} _ {i,\,j}^{m,n},0 \ right),$$
(2)
在哪里米和n是形成碱基对的两个残基的指标;我,j {1,2,3,4}表示四个伪原子的索引;\({\ hat {d}} _ {i,\,j}^{m,n} \)是两个伪原子之间的距离我和j在预测的结构中;\({{d}} _ {i,\,j}^{m,n} \)是相应的标准成对距离;氮nbpairs是该结构中所有碱基对残基的数量,并且 -是公差距离阈值。这LSS3D当两个残基形成碱基对时,核苷酸中的成对原子距离惩罚。标准成对距离的计算\({{d}} _ {i,\,j}^{m,n} \)分为两种情况:(1)当训练样本来自带有3D天然结构的PDB数据时,\({{d}} _ {i,\,j}^{m,n} \)直接来自结构,(2)当训练样本来自自缩的数据时,\({{d}} _ {i,\,j}^{m,n} \)是从相应类型的碱基对的所有PDB结构产生的统计值。这可以防止Rhofold+过度拟合伪标签,并充分利用二级结构信息。
L冲突希望该模型学会通过对原子之间的范围太短的距离来避免原子冲突。此外,我们采取损失,LLDDT,训练一位LDDT评估器,该评估者将预测的3D RNA模型作为全局回收的指标(如上所述)。目的LLDDT损失是训练一个LDDT评估器,该评估者根据地面真实结构预测预测的3D模型的LDDT。LDDT值以0.02 bin的间隔离散为50箱。一旦生成了预测的3D模型,其LDDT将以地面真实结构为基础真相PLDDT标签计算,而LDDT评估器将生成预测的PLDDT bin。横向渗透损失被用作LLDDT确定预测的LDDT是否属于地面真相箱。总体损失功能是$$ \ begin {array} {l} l = {l} _ {\ mathrm {mlm}}}+0.3 \ times {l} _ {\ mathrm {dis}}}+0.1+0.1 \ times {l}
ss}}+0.03\times {L}_{\mathrm{clash}}\\\quad+2\times
{L}_{\mathrm{FAPE}}+0.1\times {L}_{\mathrm{ss3d}}+0.01\times {L}_{\mathrm{pLDDT}}.\end{array}$$
(3)
Structure relaxation by force fields
As a preventive measure to resolve any remaining structural clashes and violations, we may relax our model predictions using a restrained energy minimization procedure, such as AMBER53and BRiQ52。Specifically, we minimized the AMBER force field using harmonic restraints, allowing the system to maintain a close relationship with its input structure.This postprediction relaxation also enforces the geometric features of phosphodiester bonds.Our empirical evidence indicates, as measured by r.m.s.d.and TM score, that while this final relaxation does not improve the model’s accuracy, it eliminates distracting stereochemical violations without compromising accuracy.
Implementation details and running time
We used the Adam optimizer with a 0.0003 learning rate for 300,000 iterations, alongside a polynomial decay scheduler with 10,000 warm-up steps and a batch size of 16. A dropout ratio of 0.1 was applied to the Rhoformer and structure modules during training. The hardware setup included a GPU cluster with 768 GB memory and eight NVIDIA A100 GPUs (80 GB each), supported by an Intel Xeon Gold 6230 central processing unit (CPU) @ 2.10 GHz with 64 cores. RhoFold+ was trained for 1,600 epochs over 300,000 iterations, taking approximately 1 week. Posttraining, the inference is rapid, with RhoFold+ predicting a structure in about 0.14 s on a single A100 GPU. For FARFAR2 benchmarking, which demands significant computational resources, a Slurm job was run on the cluster using a single central processing unit core and 8 GB memory, with execution times detailed in Supplementary Table9。
Running other baselines
In our benchmarking experiments, we obtained DeepFoldRNA, DRfold, RoseTTAFold2NA, FARFAR2 and trRosettaRNA (v1.0) from their official code repositories (either their homepages or their GitHub repositories).The authors of AlphaFold3 did not publish the code, so we used their server to perform predictions.For FARFAR2, we followed the default settings specified in the corresponding code documentation or on the server and we trained 100 models.Our benchmarking and evaluation used CASP15 natural RNA targets and newly determined single-stranded RNA structures.Following CASP15 guidelines, we collected five candidate models for each competing method to compute r.m.s.d.和Z-分数。For AlphaFold3, which generates five models per input sequence per run, we conducted one run and collected the five models.For RhoFold+, we ran it five times with different sampled MSAs, and for the other methods, we collected their five models from CASP15 website.For the newly determined single-stranded RNA structures, we ran the default configurations of RhoFold+ and other methods to produce default predictions for each sequence.For AlphaFold3, only the top model (‘model_0’) of a single run was evaluated.The input of all methods consisted solely of RNA sequences, without ions or other molecules.
报告摘要
Further information on research design is available in theNature Portfolio Reporting Summary链接到本文。
数据可用性
All data used in our work were obtained from related public datasets. We obtained all the RNA 3D structures using the data list arranged by BGSU RNA representative sets (version 2022-04-13) (http://rna.bgsu.edu/rna3dhub/nrlist/release/3.226) and downloaded them from the PDB (https://www.rcsb.org)。For pretraining our language model (RNA-FM), we downloaded the unannotated RNA sequences from RNAcentral (https://rnacentral.org/)。For RNA MSA construction, we built the database using a nucleotide database (https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nt.gz), Rfam (https://rfam.org) and RNAcentral (https://rnacentral.org) and use rMSA (https://github.com/pylelab/rMSA) for searching and construction tools. We used secondary structural information for self-distillation. For this data, we downloaded the bpRNA dataset from SPOT-RNA athttps://sparks-lab.org/server/spot-rna/, bprna-1m data fromhttps://bprna.cgrb.oregonstate.edu/and used RNAStralign, based on E2Efold available via GitHub athttps://github.com/ml4bio/e2efold。The family/type information in Rfam (https://rfam.xfam.org) was used for cross-family/type validation. For RNA-Puzzles, we downloaded native structures and submissions of other methods from GitHub athttps://github.com/RNA-Puzzles/standardized_dataset和http://www.rnapuzzles.org/results/, 分别。Similarly, CASP15 data were obtained viahttps://predictioncenter.org/casp15/index.cgi。源数据are provided with this paper.Code availability
For the RhoFold+ model, trained weights and inference scripts are available under an open-source license via GitHub at
https://github.com/ml4bio/RhoFold。RhoFold+ is also freely available as a server for academic purposes athttps://proj.cse.cuhk.edu.hk/aihlab/RhoFold/#/。Our pretrained language model (RNA-FM) and its inference pipeline can be found via GitHub athttps://github.com/ml4bio/RNA-FM。The RNA MSA search was performed by combining Infernal (http://eddylab.org/infernal/), Blastn (https://blast.ncbi.nlm.nih.gov/Blast.cgi), HMMER (http://hmmer.org) and rMSA (https://github.com/pylelab/rMSA), we also used openmm 7.7 for AMBER force field relaxation. Source codes are written under Python 3.7. We also utilized the following software for data collection, data analysis and visualization: Infernal 1.1.3 (cmbuild, cmcalibrate, cmscan, cmsearch), Cd-hit 4.8.1 (cd-hit-est), HMMER 3.3 (nhmmer), HH-suite 2.0.15, numpy 1.21.2, PyTorch 1.10.2, pandas 1.3.1, matplotlib 3.4, scikit-learn 0.24, scipy 1.7.1, biopython 1.79, PyTorch-Ignite 0.4.6 and TensorBoard 2.6.0.
参考
Mortimer, S. A., Kidwell, M. A. & Doudna, J. A. Insights into RNA structure and function from genome-wide studies.纳特。吉内特牧师。 15, 469–479 (2014).
Warner, K. D., Hajdin, C. E. & Weeks, K. M. Principles for targeting RNA with drug-like small molecules.纳特。Rev.药物发现。 17, 547–558 (2018).
Kulkarni, J. A. et al.核酸治疗的现状。纳特。纳米技术。 16, 630–643 (2021).
Sheridan, C. First small-molecule drug targeting RNA gains momentum.纳特。生物技术。 39, 6–9 (2021).
Zhao, E. M. et al. RNA-responsive elements for eukaryotic translational control.纳特。生物技术。 40, 539–545 (2022).
Liu, D., Thélot, F. A., Piccirilli, J. A., Liao, M. & Yin, P. Sub-3-Ã… cryo-em structure of RNA enabled by engineered homomeric self-assembly.纳特。方法 19, 576–585 (2022).
Xu, B. et al. Recent advances in RNA structurome.科学。中国人寿科学. 65, 1285–1324 (2022).
Rother, M., Rother, K., Puton, T. & Bujnicki, J. M. ModeRNA: a tool for comparative modeling of RNA 3D structure.核酸研究。 39, 4007–4022 (2011).
Flores, S. C., Wan, Y., Russell, R. & Altman, R. B. Predicting RNA structure by multiple template homology modeling.在过程。Pacific Symposium on Biocomputing 2010(ed. Altman, R. B. et al.) 216–227 (World Scientific, 2010).
Watkins, A. M., Rangan, R. & Das, R. Farfar2: improved de novo rosetta prediction of complex global RNA folds.结构 28, 963–976 (2020).
Wang, J., Wang, J., Huang, Y. & Xiao, Y. 3DRNA v2.0: an updated web server for RNA 3D structure prediction.国际。J.莫尔。科学。 20, 4116 (2019).
Boniecki, M. J. et al. SimRNA: a coarse-grained method for RNA folding simulations and 3D structure prediction.核酸研究。 44, e63 (2016).
Jumper, J. M. et al. Highly accurate protein structure prediction with alphafold.自然 第596章, 583–589 (2021).
Chen, X., Li, Y., Umarov, R., Gao, X. & Song, L. RNA secondary structure prediction by learning unrolled algorithms.在过程。国际学习表征会议(OpenReview, 2020);https://openreview.net/forum?id=S1eALyrYDH
Chen, J. et al.Interpretable RNA foundation model from unannotated data for highly accurate RNA structure and function predictions.预印本于https://arxiv.org/abs/2204.00300(2022)。
Townshend, R. J. et al. Geometric deep learning of RNA structure.科学 第373章, 1047–1051 (2021).
Pearce, R., Omenn, G. S. & Zhang, Y. De novo RNA tertiary structure prediction at atomic resolution using geometric potentials from deep learning.预印本于生物Rxiv https://doi.org/10.1101/2022.05.15.491755(2022)。
王,W.等人。trRosettaRNA: automated prediction of RNA 3D structure with transformer network.纳特。交流。 14, 7266 (2023).
Shen, T. et al.E2Efold-3D: end-to-end deep learning method for accurate de novo RNA 3D structure prediction.预印本于https://arxiv.org/abs/2207.01586(2022)。
Baek, M. et al. Accurate prediction of protein–nucleic acid complexes using RoseTTAFoldNA.纳特。方法 21, 117–121 (2023).
艾布拉姆森,J.等人。Accurate structure prediction of biomolecular interactions with AlphaFold 3.自然 630, 493–500 (2024).
跳,J.等人。使用 AlphaFold 进行高度准确的蛋白质结构预测。自然 第596章, 583–589 (2021).
李,Y.等人。Integrating end-to-end learning with deep geometrical potentials for ab initio RNA structure prediction.纳特。交流。 14, 5745 (2023).
Danaee, P. et al. bpRNA: large-scale automated annotation and analysis of RNA secondary structure.核酸研究。 46, 5381–5394 (2018).
Li, W. & Godzik, A. Cd-hit:用于聚类和比较大量蛋白质或核苷酸序列的快速程序。生物信息学 22, 1658–1659 (2006).
Cruz, J. A. et al. RNA-Puzzles: a CASP-like evaluation of RNA three-dimensional structure prediction.核糖核酸18 , 610–625 (2012).文章
一个 考研一个 考研中心一个 中科院一个 谷歌学术一个 Miao, Z. et al. RNA-Puzzles round II: assessment of RNA structure prediction programs applied to three large RNA structures.核糖核酸
21, 1066–1084 (2015). 文章一个
考研一个 考研中心一个 中科院一个 谷歌学术一个 Miao, Z. et al. RNA-Puzzles round III: 3D RNA structure prediction of five riboswitches and one ribozyme.核糖核酸
23, 655–672 (2017). 文章一个
考研一个 考研中心一个 中科院一个 谷歌学术一个 Miao, Z. et al. RNA-Puzzles round IV: 3D structure predictions of four ribozymes and two aptamers.核糖核酸
26, 982–995 (2020). 文章一个
考研一个 考研中心一个 中科院一个 谷歌学术一个 Magnus, M. et al. RNA-Puzzles toolkit: a computational resource of RNA 3D structure benchmark datasets, structure manipulation, and evaluation tools.核酸研究。
48, 576–588 (2020). 考研一个
中科院一个 谷歌学术一个 张,Y. 和 Skolnick,J. TM-align:基于 TM-score 的蛋白质结构比对算法。核酸研究。
33, 2302–2309 (2005). 文章一个
考研一个 考研中心一个 中科院一个 谷歌学术一个 Zhang, Y. & Skolnick, J. Scoring function for automated assessment of protein structure template quality.蛋白质
57, 702–710 (2004). 文章一个
考研一个 中科院一个 谷歌学术一个 Mariani, V., Biasini, M., Barbato, A. & Schwede, T. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests.生物信息学
29, 2722–2728 (2013). 文章一个
考研一个 考研中心一个 中科院一个 谷歌学术一个 Popenda, M. et al. Automated 3D structure composition for large RNAs.核酸研究。
40, e112 (2012). Critical assessment of techniques for protein structure prediction.Protein Structure Prediction Center
https://predictioncenter.org/casp15/index.cgi(2022)。 达斯,R.等人。Assessment of three-dimensional RNA structure prediction in CASP15.
蛋白质91 , 1747–1770 (2023).Gupta, P., Khadake, R. M., Panja, S., Shinde, K. & Rode, A. B. Alternative RNA conformations: companion or combatant.
基因13 , 1930 (2022).文章
一个 考研一个 考研中心一个 中科院一个 谷歌学术一个 Zhang, Q., Stelzer, A. C., Fisher, C. K. & Al-Hashimi, H. M. Visualizing spatially correlated dynamics that directs RNA conformational transitions.自然
450, 1263–1267 (2007). 文章一个
考研一个 中科院一个 谷歌学术一个 Ding, J. et al. Visualizing RNA conformational and architectural heterogeneity in solution.纳特。
交流。14 , 714 (2023).文章
一个 考研一个 考研中心一个 中科院一个 谷歌学术一个 Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. Rfam: an RNA family database.核酸研究。
31, 439–441 (2003). 文章一个
考研一个 考研中心一个 中科院一个 谷歌学术一个 傅,L.等人。Ufold: fast and accurate RNA secondary structure prediction with deep learning.
核酸研究。50 , e14 (2022).文章
一个 考研一个 中科院一个 谷歌学术一个 Dethoff, E. A. et al. Pervasive tertiary structure in the dengue virus RNA genome.过程。
国家科学院。科学。美国 115, 11513–11518 (2018).
Rice, G. M., Leonard, C. W. & Weeks, K. M. RNA secondary structure modeling at consistent high accuracy using differential shape.核糖核酸20 , 846–854 (2014).文章
一个 考研一个 考研中心一个 中科院一个 谷歌学术一个 Singh, J., Hanson, J., Paliwal, K. & Zhou, Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning.纳特。
交流。10 , 5407 (2019).文章
一个 考研一个 考研中心一个 谷歌学术一个 Bou-Nader, C. & Zhang, J. Structural insights into RNA dimerization: motifs, interfaces and functions.分子
25, 2881 (2020). 文章一个
考研一个 考研中心一个 中科院一个 谷歌学术一个 Trausch, J. J., Marcano-Velázquez, J. G., Matyjasik, M. M. & Batey, R. T. Metal ion-mediated nucleobase recognition by the ZTP riboswitch.化学。
生物。22 , 829–837 (2015).文章
一个 考研一个 考研中心一个 中科院一个 谷歌学术一个 Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches.生物信息学
29, 2933–2935 (2013). 文章一个
考研一个 考研中心一个 中科院一个 谷歌学术一个 Sweeney, B. A. et al. Rnacentral 2021: secondary structure integration, improved sequence search and new member databases.核酸研究。
49, D212–D220 (2021). Vaswani, A. et al.Attention is all you need.
在过程。Advances in Neural Information Processing Systems 30 (NIPS 2017)(eds Guyon, I. et al.) 5998–6008 (Curran Associates, 2017).
Rives, A. et al.生物结构和功能是通过将无监督学习扩展到 2.5 亿个蛋白质序列而产生的。过程。国家科学院。科学。美国 118, e2016239118 (2021).
Kenton, J.D.M.-W.C.& Toutanova, L.K.Bert:用于语言理解的深度双向变压器的预训练。在过程。NAACL-HLT 2019卷。1 (eds Burstein, J. et al.) 4171–4186 (Association for Computational Linguistics, 2019).
Xiong, P., Wu, R., Zhan, J. & Zhou, Y. Pairing a high-resolution statistical potential with a nucleobase-centric sampling algorithm for improving RNA model refinement.纳特。交流。 12, 2777 (2021).
Salomon-Ferrer, R., Case, D. A. & Walker, R. C. An overview of the amber biomolecular simulation package.威利跨学科。修订版计算。摩尔。科学。 3, 198–210 (2013).
致谢
This work was supported by the Chinese University of Hong Kong (CUHK; award numbers 4937025, 4937026, 5501517, 5501329, 8601603, 8601663 and SHIAE BME-p1-24 to Y.L.) and the Research Grants Council of the Hong Kong Special Administrative Region, China (Hong Kong SAR; project numbers CUHK 24204023 to Y.L., CUHK 14222922 and RGC GRF 2151185 to I.K.).Additional support was provided by the Innovation and Technology Commission of the Hong Kong SAR (project no. GHP/065/21SZ to Y.L.).J.W.was supported by a Hong Kong PhD Fellowship (award no. PF22-73180) from the Research Grants Council of the Hong Kong SAR, China and an IdeaBooster Fund (project no. IDBF24ENG06) from CUHK.The work is part of the Antibiotics-AI Project, directed by J.J.C., and supported by the Audacious Project, Flu Lab, LLC, the Sea Grape Foundation, R. Zander, H. Wyss for the Wyss Foundation, and an anonymous donor.F.W. was supported by the National Institute of Allergy and Infectious Diseases of the NIH (award no. K25AI168451).D.L.acknowledges the startup funding from ASU.We thank X.-J.Lu for guidance on interhelical twist experiments.This work was partly supported by the Shenzhen-HongKong Joint Funding Project (Category A) under grant no.SGDX20211123112401002 (to S.W.) and no.SGDX20230116092056010 (to S.W.).This project was partially supported by funds from the Focus Project of AI for Science of Comprehensive Prosperity Plan for Disciplines of Fudan University (to S.S.) and Shanghai Artificial Intelligence Laboratory (to S.S.).
道德声明
利益竞争
S.W.和 L.Z.are the co-founders of Zelixir Biotech Co. Ltd. P.Y.is a co-founder, equity holder, board member and consultant of Ultivue, Inc., Spear Bio, Inc. and Digital Biology, Inc. J.J.C.is the founding scientific advisory board chair of Integrated Biosciences.F.W. is a co-founder of Integrated Biosciences.其他作者声明没有竞争利益。
同行评审
同行评审信息
Nature Methodsthanks Hashim Al-Hashimi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.Peer reviewer reports可用。Primary Handling Editor: Arunima Singh, in collaboration with theNature Methods团队。
附加信息
Publisher’s note施普林格·自然对于已出版的地图和机构隶属关系中的管辖权主张保持中立。
Supplementary information
权利和权限
开放获取本文获得 Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License 的许可,该许可允许以任何媒介或格式进行任何非商业使用、共享、分发和复制,只要您给予原作者适当的署名即可和来源,提供知识共享许可的链接,并指出您是否修改了许可材料。根据本许可,您无权共享源自本文或其部分内容的改编材料。The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.要查看该许可证的副本,请访问http://creativecommons.org/licenses/by-nc-nd/4.0/。转载和许可
引用这篇文章

Shen, T., Hu, Z., Sun, S.
等人。Accurate RNA 3D structure prediction using a language model-based deep learning approach.自然方法(2024)。https://doi.org/10.1038/s41592-024-02487-0
已收到:
公认:
已发表:
DOI:https://doi.org/10.1038/s41592-024-02487-0