
机器学习增强免疫肽组学应用于 COVID-19 疫苗 T 细胞表位发现
作者:Caron, Etienne
介绍
由严重急性呼吸综合征冠状病毒 2 (SARS-CoV-2) 引起的 COVID-19 大流行的出现刺激了许多疫苗的快速开发1。整合 SARS-CoV-2 刺突 (S) 蛋白的疫苗的有效部署通过刺激抗体和细胞介导的免疫反应,在保护数百万人免受严重疾病方面发挥了关键作用2。然而,S蛋白在大流行期间经历了显着突变,导致SARS-CoV-2变体出现抗体逃逸机制3,4,5。这强调了促进针对保守的 SARS-CoV-2 抗原的 T 细胞免疫的重要性,这可以提供更强大的保护,预防由当前和即将出现的超突变变体引起的严重疾病6,7,8。为了实现这一目标,最近至少开发了两种 T 细胞导向疫苗 CoVac-1 和 BNT162b4,并正在进行临床试验9,10,11,12。CoVac-1是一种基于多肽的T细胞激活剂,旨在诱导广泛且持久的SARS-CoV-2 T细胞免疫,目前正进入III期临床试验(NCT04954469)9,10,11。BNT162b4 是一种基于 T 细胞的 mRNA 疫苗,编码保守的非 S 抗原,正在与 Omicron 更新的二价 BNT162b2 (NCT05541861) 结合进行临床评估12。为了加强针对未来 SARS-CoV-2 变体的下一代 T 细胞疫苗的设计,创建涵盖 HLA 分子呈现的 SARS-CoV-2 肽全谱的综合数字图谱,并详细了解他们的突变动态至关重要
13,14,15。为了实现这一目标,需要开发公正且可扩展的硬件和软件16。在这方面,基于质谱 (MS) 的免疫肽组学作为一种强大的可扩展方法脱颖而出,用于公正地鉴定 HLA 相关肽17 号,18,19。事实上,近年来,MS 越来越多地应用于表征 SARS-CoV-2 免疫肽组20,21,22,23,24,25,26,27,28,29,30。这些努力不仅揭示了传统的 SARS-CoV-2 T 细胞表位,还发现了非常规表位,包括那些由非规范翻译产生的表位,这些表位通常无法通过传统表位作图方法检测到22,23。从技术角度来看,基于 MS 的免疫肽组学涉及通过免疫亲和捕获、肽洗脱和液相色谱串联质谱 (LC-MS/MS) 采集来分离 HLA 相关肽的连续过程17 号
,31,32。随后使用蛋白质组数据库搜索引擎(例如 SEQUEST)将肽序列与从 MS/MS 获得的串联质谱进行匹配(即肽谱匹配 (PSM))33, 彗星34, MS-GF–+–35, 弗拉格女士36, 光谱矿37, 峰值38,或 Andromeda,包含在 MaxQuant 环境中39,40。这些计算工具将每个实验 MS/MS 谱与基于提供的蛋白质或肽序列数据库为每个候选肽导出的一组理论 MS/MS 谱进行比较,为每个 PSM 分配分数并报告得分最高的分数。由于每个实验 MS/MS 谱图都与至少一个肽序列相匹配,因此许多匹配都是假阳性。为了控制下游错误发现率 (FDR),诱饵肽代表来自“目标”蛋白质序列数据库的肽序列的改组或反转版本41,也用于通过数据库搜索引擎匹配实验 MS/MS 谱图。随后,目标 PSM 和诱饵 PSM 作为 PeptideProphet 等计算后处理工具的输入42,43,44和渗滤器45,46。此类工具结合了数据库搜索引擎分数以及序列和频谱属性,可用于区分目标 PSM 和诱饵 PSM。由于可能的 HLA 结合肽的搜索空间大于标准蛋白质组学实验中通常获得的肽的搜索空间,因此免疫肽组学实验中的假阳性数量往往更高47。为了尝试解决此问题,描述了其他机器学习 (ML) 和深度学习 (DL) 工具(例如 DeepRescore、Prosit、MS2Rescore、MSBooster)向 Percolator 输入文件添加功能(即肽碎片模式和保留时间),以进一步增强免疫肽组学中肽的验证48,49,50,51。最近提出了另一种在不使用蛋白质序列数据库的情况下从 MS/MS 谱图中鉴定肽的策略,以从头鉴定 HLA 肽52。
抗原加工和呈递 (APP) 的规则已被研究、评估并纳入预测算法中以预测 T 细胞表位53,54。例如,MHCflurry55,56和 NetMHCpan57,58,59是两种广泛使用的算法,它们合并来自 APP 属性的分数。MHCflurry 包含两个重要的预测因子:“抗原处理”预测因子,用于模拟 MHC 等位基因独立效应(如蛋白体裂解),以及“呈现”预测因子,将处理预测与 HLA 肽结合亲和力 (BA) 预测相结合产生综合“表现得分 (PS)”。另一方面,NetMHCpan 生成 HLA 肽结合亲和力 (BA) 和洗脱配体 (EL) 预测分数。先前的研究已将这些分数作为目标诱饵判别因素来验证免疫肽组学中的 PSM,但在保持 PSM 治疗的一致性方面面临挑战60,61,62。原则上,将这些分数更全面地纳入 PSM 置信度评估的建模过程中可以显着提高固定 FDR 下 HLA 肽识别的灵敏度和准确性,这在开发最佳疫苗的强大平台的背景下尤为重要设计。这种方法可以直接控制 FDR,这与免疫肽组学领域的流行做法相反,在免疫肽组学领域,在 FDR 估计过程之后应用分数过滤63。然而,将 APP 预测分数纳入区分目标与诱饵 PSM 的过程中,除了需要样本中表达的 HLA 等位基因的先验知识之外,还依赖于预测算法的性能。为了克服这一限制,最近的一项进展涉及序列编码器策略的创建和实施,该策略旨在学习明显表征 HLA 肽的初级序列元素64。这种方法具有更大的发现潜力,因为它考虑了成熟的和潜在的特征较少的序列基序65。然而,与 APP 预测分数不同,编码的肽序列无法轻松整合到 PSM 置信度评估软件包(例如 Percolator)中。这种限制源于 Percolator 的底层支持向量机 (SVM) 模型,该模型在容纳可变长度序列输入方面缺乏固有的灵活性。因此,需要更通用的 PSM 置信度评估方法来取代 Percolator,并利用标准 PSM 质量特征、APP 预测评分和肽氨基酸序列,以增强免疫肽组学中 HLA-I 特异性 PSM 的验证。
虽然基于 MS 的免疫肽组学显示出显着的功能,但其可及性仍然受到一定限制,阻碍了研究人员在大规模人群中直接测量 SARS-CoV-2 免疫肽组及其相关突变动态的能力16,66。相比之下,基因组测序技术的广泛应用促进了对超过 1400 万个 SARS-CoV-2 序列和 3,423 个谱系的广泛测序,包括 B.1.1.7 (alpha)、B.1.617.2 (delta)、B.1.1.617.2 (delta)、B.1.1.529 (omicron) 及其超突变变体 BA.2.86 (Pirola)67。此类大规模基因组数据已用于追踪 T 细胞表位的突变动态,以研究 SARS-CoV-2 变体的 T 细胞逃逸机制14。SARS-CoV-2 的大规模基因组测序也有助于研究 COVID-19 患者中 SARS-CoV-2 群体的宿主内变异和进化动态68。宿主内病毒的基因组异质性确实可以通过捕获宿主内单核苷酸变异(iSNV)来分析68,69。这些宿主内变异是由一小部分病毒在感染过程中随机引发的突变产生的,提供了一个突变库,塑造了病毒的快速全球进化68,69,70。已从相对较大的 COVID-19 患者群体中研究了 SARS-CoV-2 谱系的宿主内遗传多样性70,71,无论是未接种疫苗的还是已接种疫苗的个体72但将如此大规模的基因组数据与机器学习增强的免疫肽组学相结合,为针对 SARS-CoV-2 变体的疫苗设计提供信息,在很大程度上仍未得到探索。
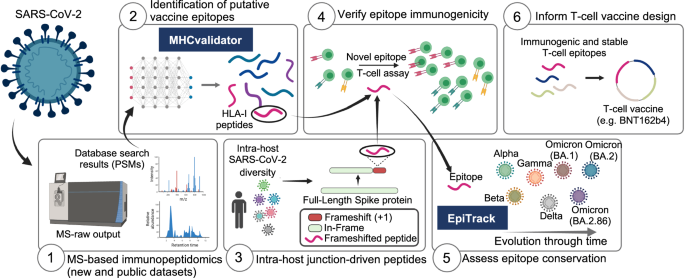
在这项工作中,我们展示了一个独特的计算框架和分析平台,为针对 SARS-CoV-2 变体的 T 细胞疫苗设计提供了宝贵的见解。该平台由六个模块组成(图1) 1):(1)基于 MS 的新的和公开的数据集的免疫肽组学,(2)基于 ML 的 MHCvalidator,(3)来自 100,512 名感染患者的大型队列中的 SARS-CoV-2 群体的宿主内基因组变异,(4) T 细胞表位免疫原性,(5) EpiTrack 用于监测 1460 万个疫苗相关 T 细胞表位的地时突变动态SARS-CoV-2 序列和 3,423 个谱系,以及 (6) 选择免疫原性和稳定的 T 细胞表位为 T 细胞疫苗设计提供信息(BNT162b4 作为本研究中测试的示例)。该分析平台适用于任何病毒。下面,我们描述了 MHCvalidator 的开发,并展示了其通过群体规模多组学数据整合和提高的免疫肽组学敏感性来促进保守 T 细胞表位疫苗候选物的公正发现的实用性。

(1) 用于数据采集的基于 MS 的免疫肽组学,(2) 用于 HLA-I 特异性 PSM 置信度评估和典型和非典型 HLA-I 病毒肽的最佳识别的 MHCvalidator,(3) SARS 的群体规模分析使用宿主内数据库的 CoV-2 蛋白质组多样性,(4) T 细胞表位免疫原性评估,(5) EpiTrack 用于跨变体表位保护的地时分析,(6) 选择免疫原性和稳定表位,为最佳 T 细胞疫苗设计提供信息。本研究分析了基于 BNT162b4 mRNA 的疫苗编码的 T 细胞表位。在 BioRender 中创建。哈梅林,D. (2024) BioRender.com/l76m979。
结果
MHCvalidator 取代 Percolator 用于免疫肽组学中的 PSM 置信度评估
MHCvalidator 能够验证基于 MS 的免疫肽组学实验中的 PSM,将数据库搜索指标和 MHC 相互作用/呈现预测因子集成到判别函数中(有关详细信息,请参阅https://github.com/CaronLab/mhc-validator和方法部分“MHC 验证器设计”)。简而言之,MHCvalidator 以多层感知器 (MLP) 神经网络验证器 (NN-validator) 作为其核心组件,并设计为在三种不同的配置中运行,这些配置可以组合起来以获得 PSM 置信度的最大增益(图 1)。 2a和补充图。 1)。来自数据库搜索引擎(例如 Comet)的搜索结果用作输入文件(PIN 或 csv)。
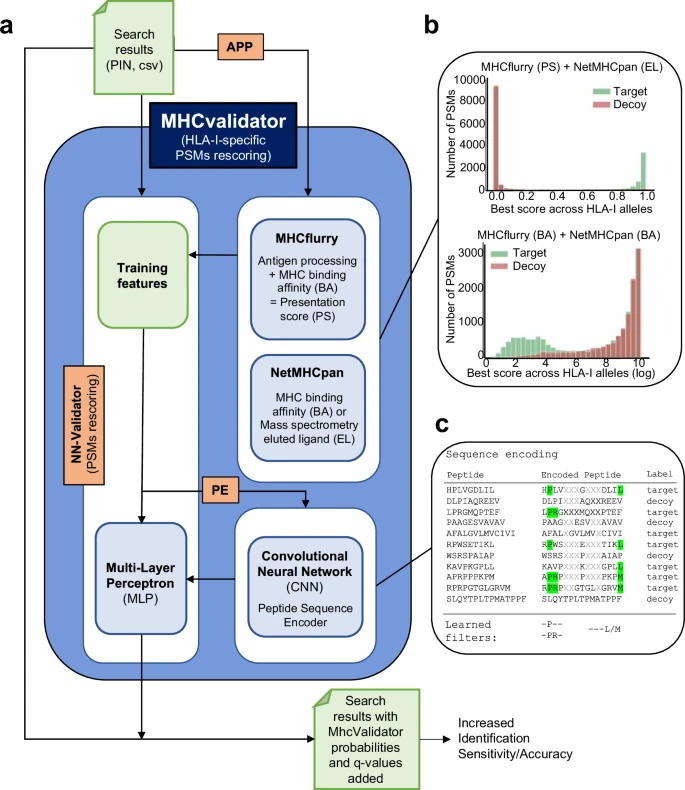
示意图说明了 MHCvalidator 的主要组件、工作流程和可能的配置。管理 MHCvalidator 配置的组件是 NN-validator、APP 和 PE(橙色)。NN-validator 代表 PSM 置信度评估的核心组件。它接受输入文件(PIN、csv/tsv)并通过多层感知器(MLP)处理训练特征;APP通过MHCflurry和NetMHCpan提供抗原加工和呈递预测评分;PE 通过卷积神经网络 (CNN) 提供编码的肽序列。乙整合 MHCflurry 和 NetMHCpan 生成的各种预测分数后目标诱饵 PSM 的示例分布。c卡通图展示了序列编码过程和基于序列组合的 PSM 重新评分的学习滤波器。使用从 JY 细胞(HLA-A*02:01;-B*07:02;-C*07:02)生成的免疫肽组数据集,我们展示了 NN 验证器在所有 FDR 低于 5% 时优于 Percolator 的性能(图
。一个 3a),特别是在低输入样本中(补充图。 1c)。除了 NN 验证器之外,MHCvalidator 还集成了另外两个关键组件:APP 预测算法(NetMHCpan 和 MHCflurry)和基于卷积神经网络 (CNN) 的肽序列编码器 (PE)(图 1)。 2a)。APP 集成了预测分数,可以清楚地显示从数据库搜索中区分目标 PSM 和诱饵 PSM 的区分能力(图 1)。 2b和补充图。 2)。PE 对肽序列进行编码,并将其作为附加数字特征直接输入到 NN 验证器的 MLP 中(图 1)。 2c)(有关详细信息,请参阅方法)。因此,MHCvalidator 实际上本质上是为了生成高置信度 HLA I 类特异性 PSM 列表而设计的。

使用 JY 细胞生成的免疫肽组学 MS 数据进行分析。一个通过 Percolator(虚线)和 MHCvalidator 的四种配置(仅限 NN-验证器:蓝色、NN-验证器和 PE:绿色、NN-验证器和 APP:橙色、NN-)在给定 FDR 下识别的 HLA-I 特异性 PSM 数量验证器、PE 和 APP:红色)。乙维恩图显示了经 MHCvalidator 和 Percolator 验证的高置信度 HLA-I 肽的数量。c使用 MixMHCp 2.1 工具从高置信度肽中提取代表性基序,这些肽由 Percolator 和 MHCvalidator(上方基序)以及 MHCvalidator(下方基序)独特识别(乙)。d代表性 MS/MS 谱图的镜像显示 Prosit 预测生成的碎片离子(底部)与 MHCvalidator 唯一识别的天然/内源肽(顶部)的比对。为每个测试的肽生成不同的值(右)。保留时间差值的分布 (e), 光谱角 (f), 人物相关性 (克) 和 Spearman 相关性 (小时) 对于使用 MHCvalidator 唯一识别的肽与通过 Percolator 和 MHCvalidator 识别的肽。源数据作为源数据文件提供。
MHCvalidator 在识别细胞系中的 HLA-I 自肽方面优于渗滤器
接下来,我们在不同的免疫肽组学实验中测试了 MHCvalidator 的配置。在 JY 细胞生成的数据集上,我们的结果表明,所有 MHCvalidator 配置在验证 HLA-I 特异性 PSM 方面均优于 Percolator(图 1)。 3a、b)。值得注意的是,“NN 验证器+PE”+“APP”配置始终在所有 PSM 级 FDR 低于 5% 的情况下提供了最有利的结果。在肽水平 FDR 1% 时,MHCvalidator(NN-validator+PE-+-APP)和 Percolator 总共验证了 4775 个高置信度 HLA-I 特异性肽,同时还验证了 1,537 个高置信度 HLA-I 肽。-MHCvalidator 独特地鉴定了特定肽(图 1) 3b和补充数据 1)。Percolator 鉴定了 3,238 个高置信度 HLA-I 特异性肽,其中只有 64 个未被 MHCvalidator 检测到。因此,MHCvalidator (NN-validator+PE–+–APP) 所鉴定的肽增加了–150%。由 MHCvalidator 唯一确定为高置信度的 PSM 显示与 JY 细胞的 A*02:01、B*07:02 和 C*07:02 相关的典型 HLA 结合基序(图 1) 3c)。为了严格验证这些独立于基序过滤的 PSM,我们采用了补充验证技术,例如 MS/MS 谱预测相似性和保留时间预测(请参阅“方法”)。对两组进行比较:(1) 由 MHCvalidator 唯一识别的 PSM,以及 (2) 由 Percolator 和 MHCvalidator 共同识别的 PSM。具体来说,我们计算了每个 PSM 的 Delta 保留时间、光谱角、Pearson 相关性和 Spearman 相关性(图 1)。 3d)。我们的研究结果表明,经 MHCvalidator 验证的 PSM 与经 Percolator 和 MHCvalidator 验证的 PSM 表现出相似的值分布(图 1)。 3e·h)。这些验证步骤证实了 MHCvalidator 独特识别的 PSM 的可靠性,确保这些肽不是误报,并为我们的研究结果增加了基于基序过滤之外的稳健性。此外,我们的分析表明 MHCvalidator 优于 DeepRescore,DeepRescore 是一种依赖于 Percolator 的 PSM 重新评分补充工具49,所有 PSM 级别的 FDR 均低于 5%(补充图 1) 3a、b)。因此,与肽验证后使用单独的 HLA 结合预测分数过滤数据相比,MHCvalidator 显着提高了固定 FDR 下肽鉴定的灵敏度。
为了比较 MHCvalidator 和 Percolator 对于大量 HLA-I 等位基因的性能,我们使用了从 71 个 HLA I 类单等位基因细胞系生成的 310 个公开可用的 MS 原始文件73。对于 Percolator 和 MHCvalidator 鉴定的总共 157,973 个高置信度 HLA-I 特异性肽 (1% FDR),其中 99% 由 Percolator 鉴定的肽也被 MHCvalidator 视为高置信度,而另外 45,388 个肽 (28%)由 MHCvalidator 唯一识别,总体性能比 Percolator 提高 1.4 倍(补充图 4a)。平均而言,与 Percolator 相比,MHCvalidator 产生的高置信度 HLA-A、-B 和 -C 特异性 PSM 分别高出约 1.34 倍、约 1.59 倍和约 1.97 倍(补充图 1)。 4b)。NN-validator+PE-+-APP 配置下的 MHCvalidator 相对于 Percolator,对实验中代表的 71 个等位基因中的 70 个实现了更高的识别灵敏度(补充图 1) 4c)。我们还观察到 PSM 验证率的增加是 HLA 等位基因依赖性的,显示中位数增加了约 1.47,HLA-C*07:01 的增加高达约 5.3 倍(补充图 1)。 4c)。有趣的是,当目标 PSM 与诱饵 PSM 的比率降低时,我们观察到 MHCvalidator 的肽识别能力比 Percolator 有所增加(补充图 1)。 4c)。这表明,当实验和数据集的质量似乎下降时,MHCvalidator 相对于 Percolator 的优势(即提高肽识别灵敏度)更大。在实验中,我们观察到高置信度 PSM 倍数增加 (MHCvalidator/Percolator) 大于 4.0,Percolator 识别的肽中有 99% 与 MHCvalidator 识别的肽重叠,而后者使肽识别率增加了 2.9 倍(3,095 比 1,076 肽),其中大多数是 HLA-C 肽并匹配其相应的结合基序(补充图。 4c)。这些结果表明,MHCvalidator 在包含较低比例的目标 PSM 的低输入样本(例如较少的肽或丰度较低的肽)中表现更有效。
为了更深入地了解 MHCvalidator 对低输入样品的性能,我们使用从 JY 细胞分离的 HLA-I 肽的两倍连续稀释(未稀释、2x、4x、8x 和 16x)生成的 MS 数据进行了评估(图。一个 4a)。使用 Comet 搜索免疫肽组学 MS 数据,然后通过 MHCvalidator(NN-validator+PE-+-APP,1% PSM 水平 FDR)或 Percolator(1% PSM 水平 FDR 和 NetMHCpan4.1 EL)评估 PSM 置信度%排名<2)。为了建立基准,我们采用了未稀释样品中 Percolator 认为高置信度的一组肽作为基准参考(图 1)。 4a)。然后,我们相对于基准测试参考评估了 MHCvalidator 和 Percolator 在每个稀释点的性能。值得注意的是,在所有稀释点,与基准参考相比,MHCvalidator 在肽鉴定方面实现了比 Percolator 更高的灵敏度,未稀释样品的改善最小(增加约 1.5 倍),而最稀释样品的改善最大(约 3.6 倍)增加)(图。 4a)。我们还观察到,MHCvalidator 在之前的稀释点始终验证了比 Percolator 更多的肽。例如,MHCvalidator 在 4 倍稀释中产生了约 3,250 个高置信度肽,而 Percolator 在 2 倍稀释中产生了约 2500 个高置信度肽。此外,MHCvalidator 认为高置信度的大多数肽与基准参考重叠,即使在 MHCvalidator 表现最佳的最稀释样品中也是如此。因此,我们的结果表明 MHCvalidator 擅长评估低输入样本中 PSM 的置信度。

直方图显示了从 JY 细胞中分离的 HLA-I 肽(x 轴)进行两倍系列稀释后,被 MHCvalidator 和 Percolator 视为高置信度的 HLA-I 特异性肽的数量(y 轴)。每个稀释度都显示了 MHCvalidator 鉴定的肽相对于 Percolator 的增加倍数。用于比较的基准参考对应于 Percolator 在未稀释样品中识别的肽 (→)。图例:在基准测试参考文献中发现由 Percolator(蓝色)和 MHCvalidator(红色)识别的肽;Percolator(浅蓝色)和 MHCvalidator(浅红色)的基准参考中未发现的高置信度肽。XCorr 值的分布 (乙)和肽长度(c)对于使用 MHCvalidator 独特发现的 PSM 与使用 Percolator 从最稀释的 JY 样品 (16x) 中发现的 PSM 进行对比。我们执行了标准独立 2 样本 t 检验,假设这些实例的总体方差相等。箱线图显示了在酵母蛋白质组中发现的 HLA-I 特异性 PSM 的数量(被认为具有高可信度)(d) 或用 Lys-C 消化的人类蛋白质组 (e)使用 Percolator 和 MHCvalidator 的四种配置(仅 NN-validator、NN-validator 和 PE、NN-validator 和 APP、以及 NN-validator 带有 PE 和 APP)。箱线图/误差线基于源自单等位基因数据集的 1550 个样本(d)。LysC 消化分析基于这些数据的子集,总共 145 个样本,是从完整的单等位基因数据集中随机选择的 (e)。箱线图以四分位数间距 (IQR) 形式给出,其中箱线从数据的第一个四分位数 (Q1) 延伸到第三个四分位数 (Q3),并在中位数处有一条线。须线从方框延伸到距方框 1.5 倍四分位数范围 (IQR) 内的最远数据点。飞行点是那些超出胡须末端的点。源数据作为源数据文件提供。
为了确定可以解释低输入样品中灵敏度差异的可能特征,我们比较了 MHCvalidator 独特识别的 PSM 和 Percolator 识别的 PSM 的质量(Xcorr 值)和肽长度,特别是在 16 倍稀释的样品中。我们的分析表明,平均而言,MHCvalidator 独特识别的肽的 Xcorr 值明显低于 Percolator 识别的肽(p≤=≤1.2'7 ≤10Ø46)(图。 4b)。然而,我们发现 MHCvalidator 独特识别的肽与 Percolator 识别的肽之间的肽长度没有显着差异(p≤=≤0.072)(图 4b)。这些结果表明差异之一在于 PSM 的质量,这表明与 Percolator 相比,MHCvalidator 对于检测识别置信度得分较低的肽具有更高的灵敏度。
为了严格评估 MHCvalidator 与 Percolator 相比的特异性,我们设计了具有挑战性的测试场景,其中根据 (1) 搜索人类免疫肽组数据集酿酒酵母/酵母蛋白质组(无酶搜索),以及 (2) 人类蛋白质组(LysC-消化搜索)。这一选择背后的基本原理源于酵母与人类相比的不同肽组成,以及与胰蛋白酶肽相比,LysC 消化的人类蛋白质组中存在更大的肽。
该测试涉及从 HLA-I 单等位基因细胞系生成的 29 个 MS 文件。该 MS 数据集针对酵母蛋白质组和 LysC 消化的人类蛋白质组进行了搜索。随后,使用 (1) MHCvalidator 的四种不同配置(NN-Validator、PE、APP、PE–+–APP)和 (2) Percolator 对结果进行置信度评估。为了确保特异性,从酵母肽数据库中删除了酵母和人类消化数据库中相同的肽序列。在检查各种 MHCvalidator 配置和 Percolator 中被视为高置信度的 PSM 分布后,我们观察到平均 <25 个 PSM 来自酵母蛋白质组搜索,因此可能构成误报。所有配置都观察到了这一点,这些配置也显示出与 Percolator 观察到的分布的视觉相似性,并且没有统计上的显着差异(p值 -= -0.7369)就被认为是高置信度的酵母 PSM 的数量而言(图 1) 4天和补充图。 3c)。LysC 消化的人类蛋白质组也产生了类似的结果(图 1)。 4e和补充图。 3d)。这些结果表明(1)APP和PE训练功能的添加不允许MHCvalidator记住特定的PSM以增加ID的数量,并且假阳性PSM的识别仍然是一个随机过程;(2) MHCvalidator 的误报识别率与 Percolator 相似。
总而言之,我们的分析表明,MHCvalidator 具有高度敏感性和特异性,在免疫肽组学实验中对 HLA-I 特异性 PSM 进行稳健的置信度评估方面优于 Percolator。
MHCvalidator 识别具有典型和非典型特性的已知和新型 MS 可检测的 SARS-CoV-2 HLA-I 肽
为了证明 MHCvalidator 在公正发现潜在 CD8+ 表位疫苗候选物方面的潜力,我们试图重新分析从 SARS-CoV-2 感染细胞生成的免疫肽组学数据23。在最初的研究中,感染了表达不同 HLA-I 等位基因组合的三种细胞系:Calu-3(HLA-A*24:02、-A*68:01、-B*07:02、-B*51:01、-C*15:02)、IHW01070(HLA-A*01:01、-A*02:01、-B*08:01、-B*40:01、-C*04:04、-C*07:01) 和 HEK293T 细胞(HLA-A*02:01、-A*03:01、-B*07:02、-C*07:02)。在这里,使用 Comet 针对人类蛋白质组和原始出版物中使用的相同 SARS-CoV-2 蛋白质组搜索相同的原始 MS 数据,然后使用 MHCvalidator (NN-validator+PE-+-APP) 或 Percolator,均为 PSM 级别 FDR 5%。值得注意的是,与原始方法(11 个肽)相比,使用 MHCvalidator 验证的 Comet 结果使可信鉴定的 HLA-I SARS-CoV-2 肽(24 个肽)的数量增加了 2.2 倍(图. 5a、b)。相比之下,被 Percolator 视为高置信度的 Comet 结果(PSM 水平 FDR 5% 和 NetMHCpan %rank<2)仅产生 6 个高置信度 HLA-I SARS-CoV-2 肽,所有这些肽也被视为高置信度- MHCvalidator 的置信度(补充图。 4天)。在未感染的细胞中未检测到任何已识别的 SARS-CoV-2 肽。然后使用 NetMHCpan 生成的预测分数将识别出的 SARS-CoV-2 肽分配给各自的 HLA-I 等位基因57和HLAthena73(图. 5b和补充图。 5)。使用体外 HLA 结合测定,大多数肽被确信分配给相应细胞系中表达的特定 HLA 等位基因,并被证实与其各自的 HLA 等位基因结合(图 1)。 5c、d)。此外,通过将合成肽的串联质谱与实验光谱进行比较,我们对 MS 可检测的 SARS-CoV-2 肽的氨基酸序列有了信心,并观察到片段离子之间的高度相关性(所有肽的平均 Pearson r = 0.9))(图。 5e和补充图。 6)。

Venn图显示了Nagler等人所描述的原始方法和MHCValidator的最佳配置(NN-VALIDATOR+PE-peâ+– pe-+â+–应用程序)。显示了重叠的肽。还指出了选择用于免疫原性实验的肽。乙表显示了MHCVALIDATOR鉴定的SARS-COV-2衍生肽的列表。源蛋白质,NetMHCPAN/HLATHENA预测评分和HLA等位基因分配在表中。在先前的研究中,MS已经检测到的肽显示了参考号。如果MS之前未检测到,则指示新的。ND:未确定。c直方图显示了确认的分配肽(Y轴)的比例,其各自的HLA-A或-b等位基因(X轴)的比例。HLA分配的预测乙),并通过体外HLA结合测定法确认。每个等位基因的肽数(分配/总数)显示在每个条的顶部。d热图说明了测得的结合亲和力(IC50NM)跨不同的HLA -A和-b等位基因,用于所有分配的肽(乙)。e镜像光谱图像显示合成与天然MHCValidated肽的MS/MS光谱中片段离子的比对。在两个MS/MS光谱之间显示了两种对免疫原性测试的代表性肽以及Pearson相关系数。f将肽分为五类。源数据作为源数据文件提供。
在Nagler等人在Nagler等人中鉴定出的11种SARS-COV-2肽中,使用我们的方法确认了7种(图。 5a)。这些肽包括stttnivtr(a*68:01,nsp3),hssgvtrel(c*15:02,nsp1)和tgsnvfqtr(a*68:01,s),以及三个核素(n)带有重叠的氨基肽的肽酸,即ritfggpsd(未分配),napritfggp(b*07:02)和apritfggp(b*07:02)(图 5b)。值得注意的是,我们还证实了非固定外肽肽GPMVLRGLIT(B*07:02)的识别,该肽源自S.iorf1/2(ORF9A),如Ribo-Seq的翻译所证明的那样74,在另一项独立研究中被MS检测到22(图 5b)。
除了上述先前报道的肽外,MS首次实际检测到一组17个SARS-COV-2肽。这些MS可检测的SARS-COV-2肽被分为五个不同的类别:(1)非典型的未框架外,(2)非经典接线驱动的,(3)规范的框架,(4)重叠,(5)> 11-mers(图 5f)。值得注意的是,我们发现了一种新型的非固定型肽RLGSPLSL(C*15:02),源自N.iorf1/2,八个新型的规范框架内肽,源自结构和非结构性SARS-SARS-COV-2个蛋白质(S,NSP3,NSP6,NSP13,NSP16),五个未分配的额外N源性肽的子集,分享了与上述相同的重叠氨基酸特性的子集和两个源自N和ORF7A的未分配的12个和13人,分别是 5b,f)。
对所有MHCVALIDATOR-CON确认的肽的深入分析都使用Ribo-Seq衍生的数据库揭示了非典型的HLA-I SARS-COV-2肽的意外类别:非经典连接驱动的肽LPYPPQILLL。该肽起源于S抗原的较短/截断版本(图 6a)由短缺失融合事件(或连接)导致的,涉及在位置去除31个核酸523594-236243(图 6b)。有趣的是,这种删除发生在类似液体的裂解部位,最近被发现并被称为与领导者无关的交界处。74。最重要的是,连接事件会产生变化的阅读框架(+1-货运),因此,非典型肽lpypqilll的翻译随后是过早的终止密码子(图。 6b)。体外HLA结合测定法表明,LPYPQILLL肽强烈结合了两个常见的HLA-B7超级型等位基因(B*07:02和B*51:01),并预测将结合相同supertype的其他常见等位基因(图。 5天,6b)。据我们所知,这是第一次报道以连接点驱动的HLA-I肽。MHCVALIDATOR一起确定了跨越规范和非典型性质的MS可检测HLA-I SARS-COV-2肽。

Wuhan-1(野生型)和截短(缺失)尖峰蛋白中的氨基酸序列。由于缺失引起的独特产生的肽序列以棕色突出显示。通过粗体和圆圈来强调LPYPQILLL肽。在 BioRender 中创建。Hamelin,D。(2024)biorender.com/K07E042。乙位置的删除(或独立领导者的交界)523594至23624-3在mRNA水平上,说明了氨基酸水平的+1帧速率。针对几个HLA-B等位基因的连接依赖性肽LPYPQILLL(橙色)的测量(非致电)或预测的(斜体)HLA结合亲和力,这些亲和力均为几个HLA-B等位基因,它们属于B7 Supertype家族。c直方图说明了托管内数据库中的患者数量,显示了100多个读取中的缺失/连接驱动的+1或+2移动率或没有移屏(框架)。在位置之间分析了删除523,623和23,693-3。d表和小提琴图指示已删除的核酸序列的长度(平均,最大和最小值),导致框架内+1或+2帧。源数据作为源数据文件提供。
由截短的S抗原编码的非典型连接驱动的B7表位的主持内分析
许多源自非典型连接的转录本已在体外记录了SARS-COV-275,76。因此,我们假设在体内SARS-COV-2感染过程中,S抗原内可能发生多个非典型连接/截断事件,以产生 +1-货物段,可能导致非典型连接肽的产生和表现在B7中+个人。为了测试这一点,我们构建了一个独特的托管内数据集,其中包括100,512个高质量的RNA库,这些数据集是从100,512例感染的COVID-19患者中测序的(有关详细信息,请参见方法)。该数据集提供了SARS-COV-2中宿主内变化的全面表示,包括在感染过程中可能在大量多样的患者中引起的突变和非典型连接。使用此独特的主宿内数据集,我们搜索了上述转录组区域附近任何长度的缺失(交界处/截断)(在S抗原的基因组位置23,623和23,693之间),可以诱导必要的边框以诱导必要的边框产生LPYPQILLL肽。我们的分析表明,在100,512名Covid-19患者中,约有1100例(〜1%)在感兴趣的区域中删除了100多个读取区域,每个删除都有100多个读取(图。 6c和补充图 7a)。值得注意的是,〜850例患者(约0.8%)显示出主要的 +1-货物段,导致非典型连接驱动的LPYPQILLL肽的编码(图。 6c和补充图 7a)。缺失长度高度可变,范围为1至100个核酸,平均缺失长度为1.3核苷酸(图。 6天和补充图 7b)。而且,我们注意到¼25%的观察到的+1移码事件可以归因于两个影响位置的核苷酸的特定缺失5 -23649(t)和5 -23657(t)(补充图。 7c)。假设我们的主席内数据集代表了SARS-COV-2感染人群,我们的结果表明¼B7的0.8%+个体可以在感染过程中表现出LPYPQILLL肽的表现。鉴于35%的人口是B7+77,我们的分析暗示了可能性¼0.3%的人口可以向CD8+T细胞呈现非典型连接驱动的S屈服。但是,进一步的实验对于在未来的研究中严格测试和验证这一观察至关重要。如果经过验证,则在感染过程中在宿主中产生的框架病毒抗原可能构成当前未开发的T细胞表位储层,并有可能在感染控制,疾病严重程度和疫苗设计中发挥作用。
MHCVALIDATOR发现的SARS-COV-2 HLA-1肽在患有COVID-19的个体中会引起CD8+T细胞反应
为了评估MS和MHCVALIDATOR检测到的HLA-I SARS-COV-2肽的免疫原性,包括上述接线驱动的B7肽,我们接下来从HLA-COVID-COVID--MATCHED-COVID-MATCHED-MATCHED COVID-MATCHEC-MONONOCAIL-ORNONOCAIL-ORSASES进行了ELISPOT测定19个疗养者[(a*68:01;n= 10),(b*07:02;n= 14),(a*68:02;n= 10),(a*02:01;n25),(b*51:01;n=â12)],并根据MS和MHCVALIDATOR验证的每种肽对IFNî³分泌进行了监测(图 7a,b)。作为阳性对照,我们将T细胞响应与S Megapool和Peppool CEF进行了比较,如所述78。我们还使用肽YLQPRTFLL(A*02:01)作为阳性对照79,80,81,82。有趣的是,非频道外肽gpmvlrglit(b*07:02),以前记录为非免疫原性22,在一个特定的b*07:02匹配的个体(〜500 sfu/10)中引起了相对有效的CD8+响应6PBMC)(图 7a)。此外,我们表明,非典型连接驱动的肽LPYPQILLL诱导了〜7%和〜42%B*07:02和B*51:01个个体的CD8+响应(图) 7a,c)。正如预期的那样,免疫主导肽YLQPRTFLL在25 HLA-A*02:01个个体(〜44%)中引起了CD8+响应。总体而言,所有肽中约有85%(在13个)结合其各自的HLA-i等位基因,并测试了免疫原性,引起了HLA匹配个体中的CD8+T细胞反应(图图) 7c)。一致地,MHCValidator识别的肽诱导阳性CD8+反应,平均频率约为25%±〜16%(图。 7天)。响应频率依赖肽依赖性,并且与预测的HLA结合亲和力显示的相关值(R)为0.568(图。 7天)。

图显示了IFNî³分泌每百万(y轴)的分泌细胞,以响应MS和MHCVALIDATOR(X轴)鉴定的肽。数据是由ELISPOT生成指定的HLA类型的。N:测试的HLA匹配的PBMC/个体数量。免疫主导肽YLQPRTFLL表示为阳性对照(+CTRL);表明对其做出反应的个体的比率(红色)。乙ELISPOT分析的代表性井图像。c饼图显示了通过ELISPOT测试的免疫原性测试的MHCVALIDATOTOTATOR-MED肽的分数。显示肽序列的表,对相应肽响应的HLA匹配个体的速率以及免疫表位数据库(IEDB)识别号(ID)。新型免疫原性肽(橙色)和先前报道的免疫原性肽(蓝色)。d图显示了ELISPOT(X轴)预测的HLA结合亲和力(Y轴)和响应频率之间的相关性。A*02:01-和A*68:01相关的肽RTIKVFTTV,表明ELISPOT和DNA-BARCODED PMHC多聚体被证明是免疫原性的。e在SARS-COV-2感染的急性期,使用了四名患者的DNA-Barcoded PMHC多聚体鉴定的肽特异性T细胞反应。确认的响应是有色的,彩色点的大小根据估计频率。指示了两名患有RTIKVFTTV和YLQPRTFLL的患者。源数据作为源数据文件提供。
为了进一步加强我们的免疫原性数据,我们将MHCValidator识别的肽与所有实验验证的反应性SARS-COV-2肽(IEDB)进行了比较。值得注意的是,用IEDB ID(2023/10/10)注释了13个MHCVALIDATOR识别的SARS-COV-2 HLA-1肽中的10个(图。 7c)。例如,在我们的研究中通过MS和ELISPOT测量的肽RTIKVFTTV的免疫原性先前是通过DNA-核编码的肽MHC多聚体测定的(图图)(图图) 7e)83。此外,在我们的研究中从未报道过三个MHCVALIDATOR鉴定的肽,在我们的研究中显示为免疫原性(图。 7c)。总之,这项研究提供了一种概念验证,即MHCVALIDATOR可以无偏见地发现来自感染细胞的免疫原性T细胞表位。
MHCVALIDATOR证实了T-Cell定向疫苗BNT162B4编码的非尖峰表位
为了展示MHCVALIDATOR在T-Cell定向疫苗的情况下的有效性,我们对DDA MS产生的免疫肽数据进行了重新分析。该数据起源于当前临床评估的BNT162B4 mRNA疫苗转染的HLA-I单相细胞系(NCT05541861)12。将彗星+MHCVALIDATOR(NN-VALIDATOR+PEâ+APP)应用于免疫肽MS数据分析,我们成功地鉴定了由BNT162B4编码的六个高信任SARS-COV-2 HLA-I肽(图。 8a和补充图 8)。预计所有高信心肽都根据其本文拓扑定位在非膜区域(补充图。 8)84。这些肽均未从相应的非转染细胞中检测到。这一发现与原始研究的结果一致,在该研究中,使用Spectrum MIL MS蛋白质组学软件v6.0实现了完全相同的肽。12,因此提供了强大的验证。例如,MHCVALIDATOR证实了肽TTDPSFLGRYM(NSP3; A*01:01)及其变异形式TTDPSFLGRY(NSP3; A*01:01)的介绍,后者报告为高度免疫性,尤其是住院患者。83。此外,MHCVALIDATOR证实了感染细胞的另一种变体[HTTDPSFLGR(NSP3; A*68:01)](图。 5)。值得注意的是,在SARS-COV-2感染的细胞或BNT162B4转导的细胞提出的16个MHCVALIDATOR识别的CD8+表位中,其中9个(56%)由NSP3编码。该观察结果表明,NSP3有可能成为下一代T细胞定向疫苗的保护性表位的主要来源。因此,MHCVALIDATOR提供了与T细胞定向的疫苗相关的免疫肽学实验中表位表现的证据,该疫苗旨在防止过度沉积的SARS-COV-2变体。

BNT162B4 mRNA疫苗的示意图。乙从SARS-COV-2感染的细胞(Orange)鉴定出的CD8+表位的综合(Gisaid,2020-2023)的突变速率;BNT162B4 mRNA疫苗(绿色);以及由9人组成的控制,跨越完整的SARS-COV-2蛋白质组(白色)。对于所有显示的表位,突变速率表示为在GISAID数据库中发现的替代表位数(每个替代表位最少10个Gisaid序列)除以该序列的GISAID序列的总数,该序列具有测序覆盖范围,即测序覆盖范围(在log10中呈现。c(底部)随着时间的推移(2020-2023)的Gisaid序列的比例(ttdpsflgry表位(Bnt162b4 mRNA疫苗,MHC-Validator识别)是未经液化的(青色)或突变的(紫色,深蓝色,浅蓝色和绿色,在下降的患病率)。此处显示的只有顶部替代表位(在> 1000个Gisaid序列中找到)。(顶)随着时间的推移,Gisaid序列的累积计数。d关注的变体(VOC)与顶级替代表位相关。颜色尺度对应于替代表位与VOC相关的Gisaid序列的数量。e顶级Ttdpsflgry替代表位的普遍性的地理图(顶部:TTDP/LSFLGRY,DELTA;底部:TTDP/SSFLGRY,OMICRON),重点是欧洲国家。颜色量表代表了每个国家产生的GISAID序列的比例,这些序列具有所讨论的替代表位,从而使国家特定的测序偏差正常化。源数据作为源数据文件提供。
表演启用疫苗与疫苗与MHCVALIDATOR的CD8+表位的地球临时保护分析
目前缺乏有关BNT162B4诱导的CD8+T细胞识别的SARS-COV-2表位突变曲线的信息。为了获得这方面的知识,我们开发了表演并分析了16个MHCValidator识别的CD8+表位的突变景观,其中包括由T细胞指导的BNT162B4疫苗编码的6个。由于SARS-COV-2的广泛基因组测序计划,可以对这些表位进行全球和时间分析。简而言之,我们通过从最终数据集中提取和翻译从GISAID中提取和翻译相关的核苷酸序列(参见方法)来汇总大流行范围的替代表位。为了深入了解每种肽的进化趋势,使用Epitrack在全球范围内和区域性地跟踪了所有相应替代肽的流行率和地理动力学。在跨越整个SARS-COV-2典型蛋白质组的一组端到端的9-Mers上重复所有分析,以评估病毒蛋白质组背景下的突变动力学。总体而言,在16个MHCValidator识别的CD8+表位中有11个,包括6个BNT162B4疫苗表位中的4个表现出大流行的可忽略不计,全球最多有1%的序列携带替代表位(图) 8b)。其余的5个肽,包括两种BNT162B4疫苗表位,其频率在4.4%至53.7%的序列之间。其中,BNT162B4疫苗表位TTDPSFLGRY和TTDPSFLGRYM特别具有临床感兴趣83。具体而言,在两个表位中都发生了两个突变,即ORF1A/NSP3 P1640L和P1640S(图。 8c e和补充图 9f)。前者在404,743个GISAID序列中被发现,在三角洲亚线序列中发现,而后者在200,770个Gisaid序列中鉴定出来,并在Omicron Sub-Lineages中发现(图。图。 8天)。预计这两种突变都可以通过其各自的HLA等位基因(a*01:01)消除两种肽的表现,这可能是由于它们发生在非锚定残基上。使用IEDB免疫原性预测指标进行预测85表明取代脯氨酸可能会对这两个表位的免疫原性产生负面影响。但是,应谨慎对待这些预测,并通过实验进行验证。其他高度突变的MHCVALIDATOR识别的表位包括B*07:02表位NapritfggP和Ranntkgsl,分别以53.7%和7.5%的序列突变(补充图。 9a和图 8b)。这些发现证明了免疫主导,与疫苗相关的表位的非平凡突变动力学,从而促进了持续监测候选T细胞疫苗内进化趋势的需求。
讨论
基于MS的免疫肽学已经成为鉴定自然呈现HLA相关肽的宝贵策略86,87,88,89。但是,这种技术的持续发展需要硬件和软件解决方案,以增强其稳健性,敏感性和特异性,最终扩大其部署和对疫苗学和免疫学的影响16。开发准确的计算框架和分析平台对于评估下一代T细胞疫苗的疗效很重要,尤其是在快速突变的SARS-COV-2的情况下。为了满足这种需求,我们开发了一种旨在公正地识别病毒T细胞表位的方法。我们方法的核心是MHCVALIDATOR,这是一种用于评估从免疫肽实验获得的PSM的ML方法。它的设计允许所有高信心PSM被认为是有趣的抗原肽,从而消除了对验证后滤波步骤的需求。在当前版本的MHCValidator中,有两个配置模块可用:应用程序和PE。后一种选项旨在解决MHC结合基序没有很好表征的方案,例如使用HLA-E等非古典MHC分子90或在具有独特MHC系统的物种中,例如蝙蝠91。在这些情况下,神经网络不能完全依靠已知的结合基序(APP模块)来进行准确的预测。PE选项与APP结合使用时,还提供了最敏感的PSM标识,具有很高的信心。因此,PE选项可以通过识别数据中的其他重要功能或信号来提供价值,从而有可能提高模型预测的准确性。此外,MHCVALIDATOR具有高度的用途,能够将各种数据属性,概率或分数作为特征,以区分false与True PSM。原则上,这种灵活性可以纳入其他数字肽特征预测,例如保留时间92,MS2光谱中的碎片强度93,94,95,离子迁移系数/碰撞横截面64,96,HLA配体演示55,73,97和免疫原性54,98进入MHCVALIDATOR的输入。此类其他功能将来可以改善MHCVALIDATOR的性能。此外,虽然当前版本的MHCVALIDATOR是为HLA-I免疫肽学实验设计的,但理论上它的适应性框架允许对HLA-II实验采用类似的方法。此外,在接受评分时,在表位近端区域作用的其他操作过程也可以集成到MHCVALIDATOR的建模过程中。自SARS-COV-2变体的背景下,这与SARS-COV-2 OMICRON BA.1 SPIKE G446S突变相关,位于同源CD8+T-cell表位的N末端之外,最近被证明可以通过三肽基肽酶II(TPPII)改善抗原加工/表现和抗病毒T细胞识别,这是一种前头膜蛋白酶介导抗原加工99。此外,MHCVALIDATOR,通过学习和纳入隐秘肽的规则和预测,包括翻译后肽剪接产生的多肽100,101,102,103,104,可以成为区分真实和假剪接肽的宝贵工具,而无需进行其他实验来验证其识别。因此,如果进一步开发和测试,则可以应用MHCVALIDATOR来继续开发专门针对免疫肽的数据库,例如SystemHC Atlas105,106,107。因此,第一个版本的MHCVALIDATOR代表了一种基于基于免疫肽学的基础数据库搜索置信度评估工具,类似于DTASELECT108和肽prophet42是在蛋白质组学创建的,后来又是渗透剂45。
在我们的研究中,MHCVALIDATOR通过三种不同的方法来验证非典型的SARS-COV-2 T细胞表位:使用合成肽对肽序列的验证,体外HLA肽结合测定法和T细胞免疫原性测定。使用ELISPOT分析在HLA型PBMC上评估了T细胞免疫原性109。我们认识到与样本量相关的局限性,包括每个肽HLA组合的10至25个数据点。根据我们在COVID-19研究中的先前经验选择了样本量,在该研究中,肽池成功刺激了PBMC,从而导致ELISPOT分析可检测到的强大的T细胞响应,并允许进行统计分析。78,110。在这项研究中,我们采用了二进制响应呈现策略,与现有文献中的方法论产生了共鸣,特别是在单个肽刺激中唤起最小反应的情况下111,112。使用免疫主导A*02:01肽YLQPRTFLL,近一半的患者(25名中的11个)表现出对刺激的积极反应,并与既定的期望保持一致。此外,如果将反应定义为正时,则确认已确定的肽的免疫原性,如果特定点数相对于阴性对照的次数超过两倍,这是某些研究中使用的阳性阈值113。因此,尽管对样本量和数据解释的二进制性质有潜在的关注,但我们认为我们的方法学是由实验环境和在现场确定的先例所支持的。我们的结果为后续研究提供了一个至关重要的基石,理想情况下将包括较大的同类人群。
在我们的研究中,一个有趣的观察结果是检测由S抗原的截短版本产生的与B7相关的SARS-COV-2非典型表位。Finkel等人首先报道了该截断的版本在连接依赖性区域。74。该连接最初是在单元线中观察到的,其数据集是独特的。在我们的研究中,我们首先想验证受感染患者中是否在体内存在完全相同的缺失,以验证Finkel的观察结果,并估算人类非经典T细胞表位的患病率。为了实现这一目标,我们构建了和询问我们的宿主内部数据库,这些数据库由成千上万的SARS-COV-2序列组成,这些序列是分离并直接从受感染的个体中进行测序的。事实证明,这些数据库在COVID-19患者中的SARS-COV-2种群中越来越强大,可以追踪宿主内部变化和进化动力学。68,69,70,114。有趣的是,我们没有发现与体外观察到的完全相同的缺失,但是我们确实观察到在S的几个区域的各种长度(1至100核酸)的大量缺失,从而导致读数框架改变。值得注意的是¼100,000名Covid-19患者,¼1100有删除,并且¼850表现出A+1-Frameshift,导致编码非典型的B7表位,以在> 100份中检测到的读数。表达水平是否足以呈现非规范的B7表位需要进一步探索。然而,很容易推测,生产截断版本的S(可能是非功能性的)可能代表一种有缺陷的核糖体产品(DRIP)形式,该形式将迅速针对蛋白酶体,以快速降解和随后的表位表现。115。鉴于存在这种移料抗原,在感染过程中可以类似地处理非典型的多肽,可能代表未开发的框架的T细胞表位来应对病毒感染。这种现象是否特定于S,其他SARS-COV-2抗原或任何病毒仍然是一个悬而未决的问题,并且需要来自不同病毒物种的宿主内部数据库。然而,了解那些未开发的非规范表位在控制感染中的重要性至关重要,因此需要进一步研究。此外,探索特定的截断事件是否与不同的临床表型相关(例如,长期相关)可以提供有价值的见解。这些知识可能为设计表型特异性疫苗的设计铺平了道路,以满足受影响个体的独特需求。
随着新的SARS-COV-2变体继续出现,最近的报告提供了令人信服的证据,证明了普遍的HLA分子呈现的免疫主导T细胞表位中的突变。116,117,118。这对T细胞的逃避具有重要意义,包括在免疫功能低下的患者中观察到的宿主内T细胞逃避119,以及诸如BA.2.86之类的超沉积变体的出现,据推测,它具有较大人群中T细胞免疫力的较高潜力120,121。人口水平的HLA等位基因的广泛多样性在某种程度上减轻了对影响T细胞表位的突变的担忧。尽管如此,我们的研究强调了对T细胞疫苗靶标的突变动态保持警惕的过程是确保未来几十年中T细胞指导疫苗持续疗效的重要过程。这对于仅包含有限数量的表位的疫苗具有特殊的意义,例如COVAC-1 T细胞疫苗的例证,该疫苗仅包括六种合成肽9。在我们的研究中,我们研究了由T细胞疫苗BNT162B4编码的MHCVALIDATOR鉴定的肽的突变动力学,尽管MRNA编码为2,257个独特的预测肽HLA-I对,但在105 HLA-I的105 HLA-i其他等位基因中检测了6肽。这种差异的潜在解释包括检测方法的敏感性不够高,或者通过细胞内蛋白酶破坏了大多数肽。值得注意的是,我们观察到疫苗蛋白的拓扑结构包括膜区域和内质网中的潜在定位。如果准确,与ER相关的降解(ERAD)途径可能在从ER膜中取代此类蛋白质中发挥作用122。但是,可以想象,此过程可能不是有效的肽表现和诱导针对疫苗靶标的强大T细胞反应的最佳选择。在这种情况下,采用各种蛋白质工程设计,成为识别和量化呈现肽的绝对丰度的宝贵工具,是一种有价值的工具123,124。未来的研究努力对于解决这些问题并进一步完善我们对疫苗设计,抗原加工途径和随后针对超节制表位的T细胞免疫反应之间的复杂相互作用的理解至关重要。
最近,已经出现了建模方法来预测未来关注的变体中的突变125。通过在T细胞表位的背景下进行进一步的完善,这些建模方法可以显着增强我们预测T细胞疫苗靶标突变概率的能力。反过来,这将使我们从大流行开始就可以预见T细胞疫苗的耐用性。本研究中提供的机器学习增强的免疫肽方法和全球表位保护分析的整合是朝这个方向迈出的重要一步。随着该框架的进一步发展,我们设想了它的潜力,可以建立和跟踪可起作用的SARS-COV-2免疫肽组的数字模型。这种模型将有助于告知下一代疫苗的制剂,不仅针对SARS-COV-2变体,而且还针对其他快速发展的病原体86,87,88。这种方法的持续完善有望增强我们在保护性T细胞指导疫苗开发中适应病毒进化的动态景观的能力。
方法
伦理
我们的研究符合所有相关的道德法规。恢复方案得到了研究MP-21-2021-3035的研究伦理委员会(REB)的批准,在Qu©Bec的五个参与中心中的每个中心中的每个参与中心中的每个中心都批准了恢复方案。Written informed consent was obtained from all participants during the recruitment period, and ongoing consent was reviewed at each subsequent visit.The sex of participants was determined by self-reporting and considered in the study design, ensuring that symptomatic and asymptomatic groups were matched for sex, age, ethnicity, and other factors.No specific gender-based analysis was conducted as the primary focus was on immune responses to SARS-CoV-2 injection.Detailed sex-disaggregated data is provided in the supplementary materials and source data files in Nantel et al.78。
Cell line and reagents
JY cell line (human lymphoblastoid B-cells) was purchased from ATCC and cultured in RPMI 1640 supplemented with 10% FBS and 1% pen/strep. Anti-human HLA-A, -B, -C (W6/32, #BE0079) was purchased from BioXcell, Polyprep chromatography column (#7311553) and Combined inhibitor EDTA-free (#A32961) from Bio-Rad and Solid phase extraction disk ultramicrospin column C18 (#SEMSS18V, 5–200 µl) from The Nest Group. 1.5 ml and 2.0 ml microcentrifuge tubes (Protein LoBind Eppendorf #022431081 and #02243100), Low retention tips Eppendorf (10 µl #2717349, 20 µl #2717351, 200 µl #2717352), acetonitrile (#A9964), trifluoroacetic acid (TFA, #AA446305Y), formic acid (#AC147930010), chaps (#22020110GM), PBS (Buph, phosphate buffer saline packs, #28372), CNBr activated sepharose 4B (#45000066) and ammonium bicarbonate (#A643-500) were purchased from Fisher.
Cell culture and immunopurification of HLA-class I peptides from JY cells
JY cells were seeded at 0.5 × 106cells/ml, incubated at 37 C° with 5% CO2and expanded to obtain 100 million cells. Cells were harvested and centrifuged at 180 × 克for a period of 5 min at room temperature. The culture medium was removed by aspiration and the cell pellets were washed gently by pipetting up and down with 5 ml of PBS and centrifuge again. After removing the PBS by aspiration, the cell pellets were stored at –80 degrees Celsius until used.Immunopurification of HLA-class I peptides
32,63。To isolate MHC class I peptides, a frozen pellet of 1 × 108cells was resuspended in 500 µL of PBS by pipetting up and down until homogenization. The volume of the cell pellet suspension was measured and transferred into a new tube 2 mL microcentrifuge tube. Equivalent volume of cell lysis buffer (1% chaps in PBS containing protease inhibitors, 1 pellet/10 mL) was added to the cell suspension (final concentration of the lysis buffer of 0.5% Chaps), followed by an incubation for 60 min at 4 °C using a rotator device and centrifugation at 18,000 × 克for 20 min at 4 °C. The cell lysis supernatant containing the MHC-peptides complexes was transferred in a new 2.0 mL microcentrifuge tube and kept on ice until used for the immunopurification. Next, 80 mg of sepharose CNBr activated beads were coupled with 2 mg of antibody. Sepharose antibody-coupled beads were incubated with the cell lysate supernatant in a 2.0 ml Low binding microcentrifuge tube overnight at 4 °C with rotation. The next day, a Bio-Rad column was installed onto a rack and pre-rinsed with 10 ml of buffer A (150 mM NaCl and 20 mM Tris–HCl pH 8). The beads-lysate mixture was transferred into the Bio-Rad column and the bottom cap was removed to discard unbound cell lysate. Beads retained in the Bio-Rad column were washed sequentially with 10 ml of buffer A (150 mM NaCl and 20 mM Tris–HCl pH 8), 10 ml of buffer B (400 mM NaCl and 20 mM Tris–HCl pH 8), 10 ml of buffer A and 10 ml of buffer C (20 mM Tris–HCl pH 8.). MHC-peptides complexes were eluted from the beads by adding 300 µl of 1% TFA, pipetting up and down 4–5 times and collecting the flowthrough. This step was repeated once and the flowthroughs were collected and combined in a new 2.0 ml tube. MHC class I peptides were desalted and eluted using a C18 column. First, the C18 column was pre-conditioned with 200 µl of (1) methanol, (2) 80% acetonitrile/0.1%TFA and (3) 0.1%TFA and spun at 1545 × 克in a fixed rotor to collect and discard the flowthroughs.Then, the MHC-peptides complexes previously collected in 600 µl of 1% TFA were loaded (3 × 200 µl) into the pre-conditioned C18 column, spun and flowthroughs were discarded.A final wash was performed with 200 µl of 0.1% TFA and spun again.Finally, the C18 column was transferred onto a 2.0 ml Eppendorf tube and MHC class I peptides were eluted with 3 × 200 µl of 28%ACN 0.1%TFA.The flowthrough containing the eluted peptides was stored at –20 degrees Celsius for MS analysis.Prior to LC-MS/MS analysis, the purified MHC class I peptides were evaporated to dryness using a vacuum concentrator with presets of temperature 45 °C, for 2 h, vacuum level: 100 mTorr and vacuum ramp: 5。
MS/MS analysis and peptide identification from JY cells for the serial dilution experiment
Vacuumed sample 1 (undiluted) was resuspended in 50 µl of 4% formic acid (FA). Twofold dilution was performed by mixing 25 µl of undiluted sample with 25 µl of 4%, and so on. Two technical replicates of 10 µl per sample were loaded and separated on a home-made reversed-phase column (150-μm i.d. by 250 mm length, Jupiter 3 µm C18 300 Å) with a gradient from 5.6 to 30% ACN-0.1% FA and a 600-nl/min flow rate on an Easy nLC-1000 connected to an Orbitrap Eclipse (Thermo Fisher Scientific). Each full MS spectrum was acquired at a resolution of 240000, an AGC of 4E5 and an injection time of 50 ms, followed by tandem-MS (MS-MS) spectra acquisition on the most abundant (Top 10) multiply charged precursor ions for a maximum of 3 s. Tandem-MS experiments were performed using higher energy collisional dissociation (HCD) at a collision energy of 34%, a resolution of 30000, an AGC of 1.5E5 and an injection time of 300 ms.
Mass spectrometry database search
Raw mass spectrometry files were converted to mzML format using ThermoRawFileParser v 1.3.4126。All data was searched using the Comet search engine (v. 2021 rev 0) with the following settings: precursor mass tolerance: 10 ppm;fragment bin size: 0.02 Da;peptide length range: 8–15 amino acids;digest enzyme: non-specific;charge state: 1–4;output format: PIN.For JY cell lines and SARS-CoV-2 infected cell lines, no fixed modifications were used and variable modifications were set to deamidation of asparagine and glutamine and oxidation of methionine.For the mono-allelic cell lines, carbamidomethylation of cystein was set as a fixed modification and variable modifications were deamidation of asparagine and glutamine and oxidation of methionine with a maximum of 3 variable modifications per peptide.The JY and mono-allelic cell line data were searched against a reference human proteome downloaded from Uniprot (downloaded 2021-05-28).The SARS-CoV-2 infection data was searched against the combined human and SARS-CoV-2 FASTA file provided in the original publication (PXD025499).For each searches, a reversed protein decoy database was appended to each FASTA file.
Validation using Percolator
Where indicated, database search results were validated using Percolator v3.05.045。Test and train FDRs were set to 0.01 and the Cpos and Cneg arguments were undefined, allowing Percolator to determine them using cross-validation.The parameters were set to output PSM results for both targets and decoys.
Validation using DeepRescore
Where indicated, samples were validated with DeepRescore, a deep learning-based algorithm for peptide identification confidence rescoring that considers predictions of peptide retention time and MS2 spectra on top of the Percolator peptide validation algorithm49。Comet database search results were used as input for DeepRescore validation.Comet searches were performed as described above with the exception that the output format was set to.pepxml instead of.pin.For DeepRescore analysis, raw files were converted to.MGF files using msConvert by ProteoWizard127(http://www.proteowizard.org/download.html)。DeepRescore was then run using the default parameters.Resulting peptides were filtered using 1% FDR cutoff and subsequently with a NetMHCpan4.1 cutoff <=2.0 to directly compare peptide quantities with Percolator and MHCvalidator.
MHCvalidator design
MHC validator can be run in three different configurations that can be combined to obtain maximum gain in confidence of PSMs: NN-validator: NN-validator represents the core component for PSMs confidence assessment. PE: PE provides encoded peptide sequences via a convolutional neural network (CNN). APP: APP provides multiple antigen processing and presentation prediction scores via MHCflurry and NetMHCpan.
Data input. The preferred input data format accepted by MHCvalidator is tab-delimited text files (TSVs). Specifically, we developed MHCvalidator using a standard Percolator input (PIN) file as the input data because of the rich feature set already present in this format. However, MHCvalidator can process any TSV-format search results. While numerical features (e.g. database search scores) are expected, the absolute minimum features required for MHCvalidator to function are peptide sequences and target-decoy labels (either as a separate feature or encoded in protein IDs with a decoy tag). MHCvalidator also provides a parser for PEPXML format search results, but PIN format is preferred as validation and testing has only been carried out using this input format.
Feature engineering. All peptide sequences present in the input data are processed using NetMHCpan and/or MHCflurry. Binding affinity and eluted ligand scores from NetMHCpan, and affinity, presentation, and processing scores from MHCflurry are added to the features present in the input data. Binding affinity predictions are transformed to a log scale before being added, with values first being clipped to a minimum value of 1e-7. NetMHCpan is run from a user-indicated installation path. Because NetMHCpan does not support the use of multiple CPUs, in order to facilitate its practical use on the typically large list of peptides, the list is split into smaller chunks which are processed concurrently. MHCflurry is automatically installed as a dependency of MHCvalidator and runs natively from within Python. For PE training, the peptide sequences are first transformed into numerical representations. In brief, similar to the sequence encoding used by MHCflurry 1.3.0, the peptides are first middle-padded with an “X†amino acid to a length of 15. They are then encoded with a BLOSUM62 matrix to which an “X†amino acid has been added (with a substitution frequency of 1 for itself and 0 for every other amino acid). These encoded sequences are the input for the PE convolutional neural network.
Artificial neural network architecture. MHCvalidator makes use of two different feed-forward artificial neural networks. The first is a multilayer-perceptron (MLP) with defaults of two hidden layers and a width of 5-times the number of input features. The second, optional network, couples the architecture of the first to a convolutional neural network that encodes peptide sequences into a numerical vector of length 6. The convolutional neural network consists of a single 1D convolutional layer with 12 filters of size 4 and stride 3, followed by a 1D max pooling layer of size 2. The output is flattened and fully connected to the output layer, which is fed to the MLP as additional training features. This model is trained simultaneously with the MLP. All hidden layers in both architectures are connected with a default dropout out of 0.5. Most model hyperparameters are exposed in the Python API and can be tuned for the dataset of interest (e.g. number of layers, layer width, dropout, batch size, number of epochs, convolutional filter size, etc.). Much like Percolator, MHCvalidator is designed with flexibility in mind. Any additional features (e.g. probabilities or scores) can be used as features and the use of the artificial neural networks is optional.
Training and predicting. When training and predicting, a K-fold cross-validation is used. The MS data is split into a variable number of sets, as defined by the user with a default of 3. Predictions made on the validation splits during the cross-validation are reported and used for calculating q-values and FDR thresholds. Because MHCvalidator can use peptide sequences or values derived from peptide sequences as training features, the splits are constructed such that duplicate peptide sequences will not be present between any training and validation sets.
Comparison of Percolator and MHCvalidator for HLA allele-specific PSMs identifications
In order to benchmark Percolator with MHCvalidator, Percolator target PSMs (1% or 5% FDR cut-off) were annotated with NetMHCpan4.1 binding predictions. Data were then filtered to keep only PSMs with an eluted ligand (EL) %Rank cut-off <=2.0, as generally performed to gain confidence in HLA-specificity15,23,63,128,129,130。Resulting PSMs were used for comparison with MHCvalidator.Different combinations of the above-described configurations (NN-validator, NN-validator+PE, NN-validator+APP, NN-validator+PE + APP) were applied to Comet outputs and compared to percolator as specified.MHCvalidator results were not filtered because the method already incorporates presentation and HLA binding affinity prediction scores into the modeling process.Peptide identifications were obtained by keeping only unique PSMs based on best score for both percolator and MHCvalidator.It is noteworthy that MHCvalidator and percolator were applied to each PIN (磷ercolator在put) file separately as opposed to applying each software to batches of files or replicates. Only unique peptides were reported across replicates, where applicable.统计和再现性All available data from immunopeptidomics experiments were utilized in this study. Statistical analyses were performed using two-sample t-tests where specified, and data visualizations, including boxplots, were created using the default settings of the Matplotlib library. All available data from immunopeptidomics experiments were utilized in this study. Statistical analyses were performed using two-sample t-tests where specified, and data visualizations, including boxplots, were created using the default settings of the Matplotlib library. No statistical method was used to predetermine sample size. No data were excluded from the analyses; the experiments were not randomized; the Investigators were not blinded to allocation during experiments and outcome assessment.Peptide clustering
Several deconvolution methods are available for analyzing binding motifs of HLA-associated peptides
131,
132
,133。We applied MixMHCp 2.1132,134to analyze 9-mer HLA-I peptides with the default settings and the number of maximum motifs set to 5. Upon completion of deconvolution, motifs were manually analyzed and assigned to JY HLA allotypes.Validation of PSMs found uniquely with MHCvalidatorPeptide-spectrum matches found uniquely with the MHCValidator software were evaluated using MS2 spectrum prediction similarity and retention time prediction. MS2 spectrum predictions were made with Prosit (https://www.nature.com/articles/s41592-019-0426-7) using the Non-tryptic 2020 HCD model. MAPDP (
https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00859
) was used to execute Prosit and compute spectrum similarity using three metrics: normalized spectral contrast angle, Pearson correlation, and Spearman correlation. Retention times were predicted using the Prosit 2019 iRT prediction model and calibrated to the experimental ones using the calibration algorithm of DeepLC (https://www.nature.com/articles/s41592-021-01301-5)。The mirror plot was drawn using spectrum-utils (0.4.2) (https://pubs.acs.org/doi/10.1021/acs.analchem.9b04884) and distribution histograms using seaborn (0.13.1) (https://joss.theoj.org/papers/10.21105/joss.03021)。SARS-CoV-2 peptide identificationTo identify SARS-CoV-2 peptides, a similar strategy to that in Nagler et al.被使用23。
Because of possible ambiguity in the identification of leucine and isoleucine residues, all isoleucine residues were substituted with leucine in the following steps.
Peptides that were assigned to both human and SARS-CoV-2 proteins were considered to be human in origin.The remaining peptides that were uniquely assigned to SARS-CoV-2 proteins were then compared with all six reading frames of the human non-coding regions (https://www.gencodegenes.org/human/release_19.html) and pseudogenes (http://www.pseudogene.org/Human/Human90.txt) used in Nagler et al. Peptides that could be attributed to human non-coding or pseudogene regions were considered to be human in origin.In vitro HLA-peptide binding assays
Peptides binding to class I HLA molecules were quantitatively measured using classical competition assays based on the inhibition of binding of a high affinity radiolabeled peptide to purified HLA molecules, as detailed elsewhere
135。Briefly, HLA molecules were purified from lysates of EBV transformed homozygous cell lines by affinity chromatography by repeated passage over Protein A Sepharose beads conjugated with the W6/32 (anti-HLA-A, -B, -C) antibody, following separation from HLA-B and -C molecules by pre-passage over a B1.23.2 (antiHLA B, C) column.Protein purity, concentration, and the effectiveness of depletion steps was monitored by SDS-PAGE and BCA assay.Peptide affinity for respective class I molecules was determined by incubating 0.1–+1 nM of radiolabeled peptide at room temperature with 1 µM to 1 nM of purified HLA in the presence of a cocktail of protease inhibitors and 1 µM B2microglobulin.Following a two-day incubation, HLA bound radioactivity was determined by capturing MHC/peptide complexes on W6/32 antibody coated Lumitrac 600 plates (Greiner Bioone, Frickenhausen, Germany).Bound cpm was measured using the TopCount (Packard Instrument Co., Meriden, CT) microscintillation counter.The concentration of peptide yielding 50% inhibition of the binding of the radiolabeled peptide was calculated.Under the conditions utilized, where [label]<[MHC] and IC50 ≥ [MHC], the measured IC50 values are reasonable approximations of the true Kd values.Each competitor peptide was tested at six different concentrations covering a 100,000-fold dose range, and in three or more independent experiments.As a positive control for inhibition, the unlabeled version of the radiolabeled probe was also tested in each experiment.
T cell immunogenicity
Subjects and samples collection: The study subjects were composed of 5 groups of previously infected health care workers (HCWs) who were recruited following a PCR-confirmed SARS-CoV-2 infection as part of the RECOVER study (n = 48). Participants were selected based on their HLA types at enrollment: (1) A68:01 (n = 10), (2) B07:02 (n = 14), (3) A68:02 (n = 10), (4) A02:01 (n = 25) and (5) B51:01 (n = 12). Some patients may share two or three different HLA types. Blood samples were collected at enrollment around 6.1 ± 2.4 months after infection into acid–citrate–dextrose tubes (ACD, BD) in each of the five participating centers in the Province of Québec, shipped to the Mother-Child Biobank at the CHU Sainte-Justine where peripheral blood mononuclear cells (PBMCs) were isolated according to standard operation procedures (SOPs) using SepMate™ tubes (Stemcell Technologies, Canada). PBMCs were cryopreserved in complete RPMI (Gibco) with 10% DMSO and stored in liquid nitrogen until used. Participants were recruited from August 17, 2020, to April 8, 2021.IFN-γ ELISpot assay. Cell-mediated immune (CMI) response was estimated by ELISpot assay. Frozen PBMCs were rapidly thawed at 37 °C and rested overnight. PBMCs were plated at 400,000 cells per well (200,000 cells for positive controls) into MultiScreenHTS-IP Filter 96-well plates (Millipore, Massachusetts, US) pre-coated with an anti-IFN-γ antibody. PBMCs were then stimulated with 10 µg/mL of single peptides identified by Mass spectrometry (MS) immunopeptidomic (STTTNIVTR, TGSNVFQTR, HTTDPSFLGR, RTIKVFTTV, APRITFGGP, NAPRITFGGP, GPMVLRGLIT, RANNTKGSL, LPYPQILLL, AADLDDFSKQLQ, HSSGVTREL, YSGVVTTV, KLPDDFTGC, YLQPRTFLL, TLNDLNETL, VPYNMRVI). PBMCs were then incubated for 48 h at 37 °C, 5% CO2. Spots were revealed using BIO-RAD Alkaline Phosphatase Conjugate Substrate Kit. The resulting ELISpots were analyzed using CTL ImmunoSpot. S5 UV Analyzer (Cellular Technology Ltd, OH). Unstimulated cells and cells stimulated with 10 μg/mL of single peptide HIV pol HLA-A*0201 (ILKEPVHGV) (10 μg/mL, JPT Peptide Technologies, Berlin, Germany) were used as negative controls and cytostim (5 uL/ml, Miltenyi, Gaithersburg, MD), spike megapool of peptides (1 μg/mL, JPT Peptide Technologies, Berlin, Germany), PepPool CEF (CD8) (1 μg/mL, JPT Peptide Technologies, Berlin, Germany) were used as positive controls. In this assay, a response was defined as positive if it was greater than or equal to the mean of negative control (response to HIV single peptide) + 3.Building a SARS-CoV-2 intra-host datasetWe compiled a comprehensive intra-host dataset, which included Illumina amplicon paired-end sequencing libraries of SARS-CoV-2 obtained during the initial two years of the COVID-19 pandemic, collected between 2020 and 2021. We ensured a representative sampling strategy across both time and geographical locations, while taking advantage of large amount of data from countries particularly involved in genomic surveillance efforts, with the United Kingdom (UK) and the United States of America (USA) contributing significantly (51% and 14% of the dataset, respectively). For each month, we randomly selected libraries based on their availability in the National Center for Biotechnology Information (NCBI): up to 5000 from the UK, up to 1000 from the USA, and up to 2000 from other global regions, resulting in a potential monthly total of 8,000 libraries, leading to the acquisition of a total of 100,512 libraries. Subsequently, each library underwent preprocessing, where Illumina sequencing adapters and low-quality reads (Phred score <20) were removed using TrimGalore! V.0.6.0. The trimmed libraries were then mapped to the SARS-CoV-2 reference genome (NC045512.2) using BWA meme v.0.7.17-r1188, resulting in BAM files. To further refine the data, we employed the iVar pipeline for primer trimming, using the ARTIC Network V3, V4, and V4.1 amplicon designs as these three kits were predominant in the sequencing centers within our dataset during the sampling period. The samtools mpileup tool, with specific parameters (-Q 20 -q 0 -B -A -d 600000), was used to generate pileup files containing read information for each BAM file. Finally, the tool pileup2base was used to convert each pileup files to the more comprehensive base file format. This process provided details such as the depth of coverage per genomic position (number of reads aligning to the position), positions of Single Nucleotide Variants (SNVs), insertions, and deletions.Identifying deletions leading to LPYPQILLL peptide
To identify all deletions leading to the generation of the LPYPQILLL CD8 + T cell epitope (frame+1), the base files generated from all SARS-CoV-2 NCBI sequencing libraries were queried using an in-house python 3.11-based algorithm. Briefly, all deletions of one or more amino acids were searched between the frame+1 stop codon directly preceding the epitope and the start of the LPYPQILLL epitope (genomic positions 23,623 and 23,693, respectively). All deletions ending prior to the stop codon as well as after the epitope were omitted. While all lengths of deletions were included in our exhaustive, comprehensive deletion analysis, only those leading to the appropriate frame shift (frame+1) were considered in the context of the LPYPQILLL peptide. To account for putative sequencing errors, three distinct thresholds were applied prior to analyses: deletions identified in at least 2 reads; 50 reads; and 100 reads.
Epitope conservation analysis using EpiTrack
A multiple sequence alignment (MSA) comprising all GISAID SARS-CoV-2 entries (n
 = 14.6 M sequences) and using Wuhan-1 (NC_045512.2) as reference was downloaded from GISAID, along with all corresponding metadata on 10/24/2023. All subsequent data processing and analyses were performed using EpiTrack (see Code Availability). GISAID entries without metadata, with incomplete year/month sampling dates, or associated with non-human hosts were omitted. The resulting dataset was used in all subsequent analyses. For all MHCvalidator-identified peptides of interest, the nucleotide sequence of the peptide was extracted from all SARS-CoV-2 sequence of the MSA (when available) and translated. The resulting sequence was defined as an “alternative peptide†if the amino acid sequence differed from that of the wuhan-1 reference sequences. The number of occurrences of all distinct alternative peptides as well as the unmutated peptide were determined. Only alternative epitopes identified in at least 10 GISAID entries were considered in subsequent analyses. As a computational control, all analyses were repeated on a set of end-to-end 9-mers spanning the entire SARS-CoV-2 canonical proteome. For all epitopes of interest (CD8+ epitopes identified from SARS-CoV-2-infected cells,n
 = 10; BNT162b4 mRNA vaccine CD8+ epitopes,n
 = 6) as well as for all control 9-mers, the epitope-specific rate of mutation was expressed as the number of alternative peptides found across the GISAID database divided by the total number of GISAID sequences for which the epitope had sequencing coverage.
报告摘要Further information on research design is available in the Nature Portfolio Reporting Summarylinked to this article.数据可用性The mass spectrometry JY immunopeptidomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifierPXD052187。
Other mass spectrometry datasets for this study were acquired from the following public repositories: mono-allelic cell line data - MassIVE: MSV000080527 (
https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=a67baac756f5421faf51c5d4bac3005f);BNT162b4 data - MassIVE repository: MSV000091008 (
https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=88caf6334d494a72b3c8d5e9988a64e6
);SARS-CoV-2 infections - PRIDE repository:PXD025499。一个 源数据随本文一起提供。代码可用性MHCvalidator is available on the CaronLab GitHub page:https://github.com/CaronLab/mhc-validator。Instructions on how to run MHCvalidator in its different configurations are explained in this page.The scripts pertaining to the intra-host deletion analyses, along with the list of NCBI libraries used, as well as the mutational dynamics analyses are available on the HussinLab GitHub page:
https://github.com/HussinLab/EpiTrack
。Source code are also available on Zenodo: MHCvalidator (https://zenodo.org/records/13736549) and EpiTrack (https://zenodo.org/records/13738788)136。参考Le, T. T. et al. The COVID-19 vaccine development landscape.纳特。
Rev.药物发现。
19, 305–306 (2020). 文章一个
谷歌学术一个 Watson, O. J. et al. Global impact of the first year of COVID-19 vaccination: A mathematical modelling study.柳叶刀感染。
迪斯。22 , 1293–1302 (2022).文章
一个 中科院一个 考研一个 考研中心一个 谷歌学术一个 曹,Y.等人。Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies.
自然602 , 657–663 (2022).文章
一个 ADS一个 中科院一个 考研一个 谷歌学术一个 王,Q.等人。Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants.
细胞186 , 279–286.e8 (2023).文章
一个 中科院一个 考研一个 考研中心一个 谷歌学术一个 简,F.等人。Further humoral immunity evasion of emerging SARS-CoV-2 BA.4 and BA.5 subvariants.
柳叶刀感染。迪斯。 22, 1535–1537 (2022).
Sette, A. & Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19.细胞 184, 861–880 (2021).
Wherry, E. J. & Barouch, D. H. T cell immunity to COVID-19 vaccines.科学 第377章, 821–822 (2022).
Diniz, M. O., Maini, M. K. & Swadling, L. T cell control of SARS-CoV-2: When, which, and where?塞明。免疫学。 70, 101828 (2023).
Heitmann, J. S. et al. A COVID-19 peptide vaccine for the induction of SARS-CoV-2 T cell immunity.自然 601, 617–622 (2021).
Tandler, C. et al. Long-term efficacy of the peptide-based COVID-19 T cell activator CoVac-1 in healthy adults.国际。J.感染。迪斯。 139, 69–77 (2023).
Heitmann, J. S. et al. Phase I/II trial of a peptide-based COVID-19 T-cell activator in patients with B-cell deficiency.纳特。交流。 14, 5032 (2023).
Arieta, C. M. et al. The T-cell-directed vaccine BNT162b4 encoding conserved non-spike antigens protects animals from severe SARS-CoV-2 infection.细胞 186, 2392–2409 (2023).
Nathan, A. et al. Structure-guided T cell vaccine design for SARS-CoV-2 variants and sarbecoviruses.细胞 184, 4401–4413.e10 (2021).
Hamelin, D. J. et al. The mutational landscape of SARS-CoV-2 variants diversifies T cell targets in an HLA supertype-dependent manner.细胞系统。 13, 143–157 (2021).
Caron, E. et al. An open-source computational and data resource to analyze digital maps of immunopeptidomes.埃莱夫 4, e07661 (2015).
Kapoor, S., Maréchal, L., Sirois, I. & Caron, É. Scaling up robust immunopeptidomics technologies for a global T cell surveillance digital network.J.Exp。医学。 221, e20231739 (2024).
Caron, E. et al. Analysis of major histocompatibility complex (MHC) immunopeptidomes using mass spectrometry.摩尔。细胞蛋白质组。 14, 3105–3117 (2015).
Kubiniok, P. et al. Understanding the constitutive presentation of MHC class I immunopeptidomes in primary tissues.科学 25, 103768 (2022).
Kovalchik, K., Hamelin, D. & Caron, E. Generation of HLA allele-specific spectral libraries to identify and quantify immunopeptidomes by SWATH/DIA-MS.方法分子。生物。 2420, 137–147 (2021).
Parker, R. et al. Mapping the SARS-CoV-2 spike glycoprotein-derived peptidome presented by HLA class II on dendritic cells.细胞代表。 35, 109179 (2021).
Knierman, M. D. et al. The human leukocyte antigen class II immunopeptidome of the SARS-CoV-2 spike glycoprotein.细胞代表。 33, 108454 (2020).
Weingarten-Gabbay, S. et al. Profiling SARS-CoV-2 HLA-I peptidome reveals T cell epitopes from out-of-frame ORFs.细胞 184, 3962–3980 (2021).
Nagler, A. et al. Identification of presented SARS-CoV-2 HLA class I and HLA class II peptides using HLA-peptidomics.细胞代表。 35, 109305 (2021).
Pan, K. et al. Mass spectrometric identification of immunogenic SARS-CoV-2 epitopes and cognate TCRs.过程。国家。阿卡德。科学。美国 118, e2111815118 (2021).
Gomez-Zepeda, D. et al. Thunder-DDA-PASEF enables high-coverage immunopeptidomics and is boosted by MS2Rescore with MS2PIP timsTOF fragmentation prediction model.纳特。交流。 15, 2288 (2023).
Weingarten-Gabbay, S. et al. The HLA-II immunopeptidome of SARS-CoV-2.细胞代表。 43, 113596 (2023).
Purcell, A. et al. Mapping the immunopeptidome of seven SARS-CoV-2 antigens across common HLA haplotypes.https://doi.org/10.21203/rs.3.rs-3564516/v1(2023)。
El-Baky, N. A., Amara, A. A. & Redwan, E. M. HLA-I and HLA-II peptidomes of SARS-CoV-2: A review.疫苗 11, 548 (2023).
Nelde, A. et al. Increased soluble HLA in COVID-19 present a disease-related, diverse immunopeptidome associated with T cell immunity.科学 25, 105643 (2022).
Becerra-Artiles, A. et al. Immunopeptidome profiling of human coronavirus OC43-infected cells identifies CD4 T-cell epitopes specific to seasonal coronaviruses or cross-reactive with SARS-CoV-2.PLoS Pathog。 19, e1011032 (2023).
Purcell, A. W., Ramarathinam, S. H. & Ternette, N. Mass spectrometry–based identification of MHC-bound peptides for immunopeptidomics.纳特。协议。 14, 1687–1707 (2019).
Sirois, I., Isabelle, M., Duquette, J. D., Saab, F. & Caron, E. Immunopeptidomics: Isolation of mouse and human MHC class I- and II-associated peptides for mass spectrometry analysis.J. 维斯。经验值。https://doi.org/10.3791/63052(2021)。Eng, J. K., McCormack, A. L. & Yates, J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database.
J. Am.苏克。质谱。 5, 976–989 (1994).
Eng, J. K., Jahan, T. A. & Hoopmann, M. R. Comet: An open-source MS/MS sequence database search tool.蛋白质组学 13, 22–24 (2012).
Kim, S. & Pevzner, P. A. MS-GF+ makes progress towards a universal database search tool for proteomics.纳特。交流。 5, 5277 (2014).
Kong, A. T., Leprevost, F. V., Avtonomov, D. M., Mellacheruvu, D. & Nesvizhskii, A. I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics.纳特。方法 14, 513–520 (2017).
Muntel, J. et al. Surpassing 10000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy.摩尔。组学 15, 348–360 (2019).
Xin, L. et al. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics.纳特。交流。 13, 3108 (2022).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification.纳特。生物技术。 26, 1367–1372 (2008).
Tyanova, S.、Temu, T. 和 Cox, J. 用于基于质谱的鸟枪蛋白质组学的 MaxQuant 计算平台。纳特。协议。 11, 2301–2319 (2016).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry.纳特。方法 4, 207–214 (2007).
Keller, A., Nesvizhskii, A. I., Kolker, E. & Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search.肛门。化学。 74, 5383–5392 (2002).
Shteynberg, D. et al. iProphet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates.摩尔。细胞蛋白质组学 10, M111.007690 M111.007690 (2011).
Ma, K., Vitek, O. & Nesvizhskii, A. I. A statistical model-building perspective to identification of MS/MS spectra with PeptideProphet.BMC 生物信息。 13, S1 (2012).
Käll, L., Canterbury, J. D., Weston, J., Noble, W. S. & MacCoss, M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets.纳特。方法 4, 923–925 (2007).
The, M., MacCoss, M. J., Noble, W. S. & Käll, L. Fast and accurate protein false discovery rates on large-scale proteomics data sets with Percolator 3.0.J. Am.苏克。质谱。 27, 1719–1727 (2016).
Jeong, K., Kim, S. & Bandeira, N. False discovery rates in spectral identification.BMC Bioinform。13, S2 (2012).
Declercq, A. et al. MS2Rescore: Data-driven rescoring dramatically boosts immunopeptide identification rates.摩尔。细胞蛋白质组。 21, 100266 (2022).
Li, K., Jain, A., Malovannaya, A., Wen, B. & Zhang, B. DeepRescore: Leveraging deep learning to improve peptide identification in immunopeptidomics.蛋白质组学1900334.https://doi.org/10.1002/pmic.201900334(2020)。
Wilhelm, M. et al.深度学习提高了基于质谱的免疫肽组学的灵敏度。纳特。交流。 12, 3346 (2021).
Yang, K. L. et al. MSBooster: improving peptide identification rates using deep learning-based features.纳特。交流。 14, 4539 (2023).
廖,H.等人。MARS an improved de novo peptide candidate selection method for non-canonical antigen target discovery in cancer.纳特。交流。 15, 661 (2024).
Peters, B., Nielsen, M. & Sette, A. T cell epitope predictions.免疫学年鉴。 38, 123–145 (2020).
Albert, B. A. et al. Deep neural networks predict class I major histocompatibility complex epitope presentation and transfer learn neoepitope immunogenicity.纳特。马赫。英特尔。 5, 861–872 (2023).
O’Donnell, T. J., Rubinsteyn, A. & Laserson, U. MHCflurry 2.0: Improved pan-allele prediction of MHC class I-presented peptides by incorporating antigen processing.细胞系统。 11, 42–48.e7 (2020).
O’Donnell, T. J. et al. MHCflurry: Open-source class I MHC binding affinity prediction.细胞系统。 7, 129–132.e4 (2018).
Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data。核酸研究。 48, W449–W454 (2020).
Nilsson, J. B. et al. Accurate prediction of HLA class II antigen presentation across all loci using tailored data acquisition and refined machine learning.科学。副词。 9, eadj6367 (2023).
Jurtz, V. et al. NetMHCpan-4.0: Improved Peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data.J.免疫学。 199, 3360–3368 (2017).
Andreatta, M. et al. MSâ€Rescue: A computational pipeline to increase the quality and yield of immunopeptidomics experiments.蛋白质组学 19, 1800357 (2019).
Granados, D. P. et al. Impact of genomic polymorphisms on the repertoire of human MHC class I-associated peptides.纳特。交流。 5, 3600 (2014).
Bichmann, L. et al. MHCquant: Automated and reproducible data analysis for immunopeptidomics.J. Proteome Res。18, 3876–3884 (2019).文章
一个 中科院一个 考研一个 谷歌学术一个 Kovalchik, K. A. et al. MhcVizPipe: A quality control software for rapid assessment of small- to large-scale immunopeptidome data sets.摩尔。
细胞蛋白质组。21 , 100178 (2021).文章
AlphaPeptDeep: a modular deep learning framework to predict peptide properties for proteomics.纳特。交流。 13, 7238 (2022).
Falk, K., Rötzschke, O., Stevanovic, S., Jung, G. & Rammensee, H.-G. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules.自然 第351章, 290–296 (1991).
VizcaÃno, J. A. et al. The Human Immunopeptidome Project: a roadmap to predict and treat immune diseases.摩尔。细胞蛋白质组。 19, 31–49 (2020).
杨,S.等人。Antigenicity and infectivity characterisation of SARS-CoV-2 BA.2.86.柳叶刀感染。迪斯。 23, e457–e459 (2023).
王,Y.等人。Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients.基因组医学。 13, 30 (2021).
李,J.等人。Two-step fitness selection for intra-host variations in SARS-CoV-2.细胞代表。 38, 110205 (2022).
Pathak, A. K. et al. Spatio-temporal dynamics of intra-host variability in SARS-CoV-2 genomes.核酸研究。 50, 1551–1561 (2022).
刘,Y.等人。Rescuing low frequency variants within intra-host viral populations directly from Oxford Nanopore sequencing data.纳特。交流。 13, 1321 (2022).
顾,H.等人。Within-host genetic diversity of SARS-CoV-2 lineages in unvaccinated and vaccinated individuals.纳特。交流。 14, 1793 (2023).
萨尔基佐娃,S.等人。大型肽组数据集改进了大多数人群的 HLA I 类表位预测。纳特。生物技术。 38, 199–209 (2020).
Finkel, Y. et al. The coding capacity of SARS-CoV-2.自然 第589章, 125–130 (2021).
Davidson, A. D. et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein.基因组医学。12, 68 (2020).
金,D.等人。The architecture of SARS-CoV-2 transcriptome.细胞 181, 914–921.e10 (2020).
Francisco, R. et al. HLA supertype variation across populations: new insights into the role of natural selection in the evolution of HLA-A and HLA-B polymorphisms.免疫遗传学 67, 651–663 (2015).
Nantel, S. et al. Symptomatology during previous SARS-CoV-2 infection and serostatus before vaccination influence the immunogenicity of BNT162b2 COVID-19 mRNA vaccine.前免疫学。 13, 930252 (2022).
Ferretti, A. P. et al. Unbiased screens show CD8+ T cells of COVID-19 patients recognize shared epitopes in SARS-CoV-2 that largely reside outside the spike protein.免疫 53, 1095–1107.e3 (2020).
Shomuradova, A. S. et al. SARS-CoV-2 epitopes are recognized by a public and diverse repertoire of human T cell receptors.免疫 56, 1245–1257 (2020).
Schulien, I. et al. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells.纳特。医学 27, 78–85 (2021).
Alter, G. et al. Immunogenicity of Ad26.COV2.S vaccine against SARS-CoV-2 variants in humans.自然 第596章, 268–272 (2021).
Saini, S. K. et al. SARS-CoV-2 genome-wide T cell epitope mapping reveals immunodominance and substantial CD8+ T cell activation in COVID-19 patients.科学。免疫学。 6, eabf7550 (2021).
Omasits, U., Ahrens, C. H., Müller, S. & Wollscheid, B. Protter: interactive protein feature visualization and integration with experimental proteomic data.生物信息学 30, 884–886 (2014).
Calis, J. J. A. et al. Properties of MHC class I presented peptides that enhance immunogenicity.PLoS 计算生物学。 9, e1003266 (2013).
Mayer, R. L. et al. Immunopeptidomics-based design of mRNA vaccine formulations against Listeria monocytogenes.纳特。交流。 13, 6075 (2022).
Mayer, R. L. & Impens, F. Immunopeptidomics for next-generation bacterial vaccine development.微生物趋势 29, 1034–1045 (2021).
Ovsyannikova, I. G., Johnson, K. L., Bergen, H. R. & Poland, G. A. Mass spectrometry and peptide-based vaccine development.临床。医药。瑟尔。 82, 644–652 (2007).
Bettencourt, P. et al. Identification of antigens presented by MHC for vaccines against tuberculosis.NPJ 疫苗 5, 2 (2020).
D’Souza, M. P. et al. Casting a wider net: Immunosurveillance by nonclassical MHC molecules.PLoS Pathog。 15, e1007567 (2019).
曲,Z.等人。Structure and peptidome of the bat MHC cass I molecule reveal a novel mechanism leading to high-affinity peptide binding.J.免疫学。 第202章, 3493–3506 (2019).
Wen, B., Li, K., Zhang, Y. & Zhang, B. Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis.纳特。交流。 11, 1759 (2020).
Gessulat,S.等人。Prosit:通过深度学习对肽串联质谱进行全蛋白质组预测。纳特。方法 16, 509–518 (2019).
Degroeve, S. & Martens, L. MS2PIP: A tool for MS/MS peak intensity prediction.生物信息学 29, 3199–3203 (2013).
Zhou, X.-X.等人。pDeep:通过深度学习预测肽的 MS/MS 谱。肛门。化学。 89, 12690–12697 (2017).
迈尔,F.等人。Deep learning the collisional cross sections of the peptide universe from a million experimental values.纳特。交流。 12, 1185 (2021).
Racle, J. et al. Machine learning predictions of MHC-II specificities reveal alternative binding mode of class II epitopes.免疫 56, 1359–1375 (2023).
Müller, M. et al. Machine learning methods and harmonized datasets improve immunogenic neoantigen prediction.免疫 56, 2650–2663.e6 (2023).
Motozono, C. et al. The SARS-CoV-2 Omicron BA.1 spike G446S mutation potentiates antiviral T-cell recognition.纳特。交流。 13, 5440 (2022).
Hanada, K., Yewdell, J. W. & Yang, J. C. Immune recognition of a human renal cancer antigen through post-translational protein splicing.自然 第427章, 252–256 (2004).
Delong, T. et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion.科学 第351章, 711–714 (2016).
Paes, W. et al. Contribution of proteasome-catalyzed peptide cis-splicing to viral targeting by CD8+ T cells in HIV-1 infection.过程。国家科学院。科学。美国 116, 244748–24759 (2019).
Tran, M. T. et al. T cell receptor recognition of hybrid insulin peptides bound to HLA-DQ8.纳特。交流。 12, 5110 (2021).
Saab, F. et al. RHybridFinder: An R package to process immunopeptidomic data for putative hybrid peptide discovery.星星。协议。 2, 100875 (2021).
Shao, W. et al. The SysteMHC Atlas project.核酸研究。 46, D1237–D1247 (2017).
黄X.等人。The SysteMHC Atlas v2.0, an updated resource for mass spectrometry-based immunopeptidomics.核酸研究。 52, D1062–D1071 (2024).
Shao, W., Caron, E., Pedrioli, P. & Aebersold, R.方法分子。生物。 2120, 173–181 (2020).
Tabb, D. L., McDonald, W. H. & Yates, J. R. DTASelect and contrast: Tools for assembling and comparing protein identifications from Shotgun proteomics.J.蛋白质组研究。 1, 21–26 (2002).
Racine, É.等人。The REinfection in COVIDâ€19 Estimation of Risk (RECOVER) study: Reinfection and serology dynamics in a cohort of Canadian healthcare workers.Influenza Other Resp. 16, 916–925 (2022).
Nantel, S. et al. Comparison of Omicron breakthrough infection versus monovalent SARS-CoV-2 intramuscular booster reveals differences in mucosal and systemic humoral immunity.粘膜免疫学。 17 号, 201–210 (2024).
赵,J.等人。SARS-CoV-2 specific memory T cell epitopes identified in COVID-19-recovered subjects.病毒研究。 304, 198508 (2021).
Skelly, D. T. et al. Two doses of SARS-CoV-2 vaccination induce robust immune responses to emerging SARS-CoV-2 variants of concern.纳特。交流。 12, 5061 (2021).
Mizukoshi, E. et al. Peptide vaccine-treated, long-term surviving cancer patients harbor self-renewing tumor-specific CD8+ T cells.纳特。交流。 13, 3123 (2022).
Fournelle, D. et al. Intra-host viral populations of SARS-CoV-2 in immunosuppressed patients with hematologic cancers.生物Rxiv2022.10.19.512884.https://doi.org/10.1101/2022.10.19.512884(2022)。
Dersh, D., Hollý, J. & Yewdell, J. W. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion.纳特。免疫学牧师。 21, 116–128 (2020).
Naranbhai, V. et al. T cell reactivity to the SARS-CoV-2 Omicron variant is preserved in most but not all individuals.细胞 185, 1041–1051.e6 (2022).
Motozono, C. et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity.细胞宿主微生物 29, 1124–1136.e11 (2021).
Dolton, G. et al. Emergence of immune escape at dominant SARS-CoV-2 killer T cell epitope.细胞 185, 2936–2951.e19 (2022).
Stanevich, O. V. et al. SARS-CoV-2 escape from cytotoxic T cells during long-term COVID-19.纳特。交流。 14, 149 (2023).
Sette, A., Sidney, J. & Grifoni, A. Pre-existing SARS-2-specific T cells are predicted to cross-recognize BA.2.86.细胞宿主微生物 32, 19–24.e2 (2023).
Müller, T. R. et al. Memory T cells effectively recognize the SARS-CoV-2 hypermutated BA.2.86 variant.细胞宿主微生物 32, 156–161.e3 (2024).
Meusser, B., Hirsch, C., Jarosch, E. & Sommer, T. ERAD: the long road to destruction.纳特。细胞生物学。 7, 766–772 (2005).
Purcell, A. W., Croft, N. P. & Tscharke, D. C. Immunology by numbers: quantitation of antigen presentation completes the quantitative milieu of systems immunology!电流。意见。免疫学。 40, 88–95 (2016).
Stutzmann, C. et al. Unlocking the potential of microfluidics in mass spectrometry-based immunopeptidomics for tumor antigen discovery.Cell Rep. Methods 3, 100511 (2023).
Thadani, N. N. et al. Learning from prepandemic data to forecast viral escape.自然 622, 818–825 (2023).
Hulstaert, N. et al. ThermoRawFileParser: Modular, scalable, and cross-platform RAW file conversion.J.蛋白质组研究。 19, 537–542 (2020).
Adusumilli, R. & Mallick, P. Proteomics, Methods and Protocols.方法分子。生物。 1550, 339–368 (2017).
Kraemer, A. I. et al. The immunopeptidome landscape associated with T cell infiltration, inflammation and immune editing in lung cancer.纳特。癌症 4, 608–628 (2023).
Kina, E. et al. Breast cancer immunopeptidomes contain numerous shared tumor antigens.J.克林。调查。134。https://doi.org/10.1172/jci166740(2023)。Courcelles, M. et al. MAPDP: a cloud-based computational platform for immunopeptidomics analyses.J. Proteome Res
19, 1873–1881 (2020). 文章一个
中科院一个 考研一个 谷歌学术一个 Andreatta, M., Alvarez, B. & Nielsen, M. GibbsCluster: Unsupervised clustering and alignment of peptide sequences.核酸研究
。https://doi.org/10.1093/nar/gkx248(2017)。Bassani-Sternberg, M. & Gfeller, D. Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide–HLA interactions.J.免疫学。
197, 2492–2499 (2016). 文章一个
中科院一个 考研一个 谷歌学术一个 Shahbazy, M. et al. MHCpLogics: An interactive machine learning-based tool for unsupervised data visualization and cluster analysis of immunopeptidomes.简短的。
生物信息。25 , bbae087 (2024).文章
一个 考研一个 考研中心一个 谷歌学术一个 Gfeller, D. et al. The length distribution and multiple specificity of naturally presented HLA-I ligands.J.免疫学。
201, 3705–3716 (2018). 文章一个
中科院一个 考研一个 谷歌学术一个 Sidney, J. et al.通过凝胶过滤或单克隆抗体捕获测量 MHC/肽相互作用。
电流。协议。免疫学 18, Unit 18.3 (2023).
Kovalchik, K. A., et al. Machine learning-enhanced immunopeptidomics advances T-cell epitope discovery for COVID-19 vaccines, Zenodo,https://doi.org/10.5281/zenodo.13736548(MHCvalidator) andhttps://doi.org/10.5281/zenodo.13738767(EpiTrack), 2024.
致谢
This work was supported by start-up funding from Yale School of Medicine (EC) as well as funding from the Fonds de recherche du Québec – Santé (FRQS) (EC), the Cole Foundation (EC), CHU Sainte-Justine and the Charles-Bruneau Foundations (EC), Canada Foundation for Innovation (EC), the National Sciences and Engineering Research Council (NSERC) (#RGPIN-2020-05232) (EC), the Canadian Institutes of Health Research (CIHR) (#174924, #172712) (EC, JGH), the CIHR Coronavirus Variants Rapid Response Network (CoVaRR-Net) (#ARR-175622) (EC) and a NSERC Discovery Grant (MLA). IRIC proteomics facility is a Genomics Technology platform funded in part by the Canadian Government through Genome Canada (PT). IEDB is supported by 75N93019C00001/AI/NIAID NIH HHS/United States (AS). KK is a recipient of IVADO’s postdoctoral scholarship (#4879287150) (KK). DJH is a recipient of the Hydro-Quebec doctoral and FRQS fellowships (DJH). JGH is FRQS Junior 2 research scholar (JGH). We gratefully acknowledge the GISAID Initiative and the generous contribution of all data contributors to both GISAID and NCBI, including the authors, their laboratories that collect the specimens and generated the genetic sequence and metadata on which part of this research is based. This work was completed thanks to computational resources provided by Digital Research Alliance of Canada, particularly Narval and Beluga clusters.
道德声明
利益竞争
E.C. and I.S. are co-founders of Neomabs Biotechnologies Inc. PT is a co-founder of Epitopea. A.S. is a consultant for Alcimed, Gritstone, Darwin Health, EmerVax, Gilead Sciences, Guggenheim Securities, Link University, RiverVest Venture Partners, and Arcturus. La Jolla Institute for Immunology has filed for patent protection for various aspects of T cell epitope and vaccine design work. All other authors declare no competing interests.
Inclusion & Ethics Statement
This research prioritizes inclusivity by engaging diverse populations, particularly underrepresented groups. All participants provided informed consent, and their confidentiality was safeguarded in accordance with ethical guidelines. The contributions of all team members were acknowledged, and potential biases were actively addressed. We aim to conduct research that advances knowledge while respecting the rights and dignity of all individuals involved.
同行评审
同行评审信息
自然通讯thanks David Gfeller and the other, anonymous, reviewers for their contribution to the peer review of this work.同行评审文件可用。
附加信息
Publisher’s note施普林格·自然对于已出版的地图和机构隶属关系中的管辖权主张保持中立。
补充资料
权利和权限
开放获取本文获得 Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License 的许可,该许可允许以任何媒介或格式进行任何非商业使用、共享、分发和复制,只要您给予原作者适当的署名即可和来源,提供知识共享许可的链接,并指出您是否修改了许可材料。根据本许可,您无权共享源自本文或其部分内容的改编材料。The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.要查看该许可证的副本,请访问http://creativecommons.org/licenses/by-nc-nd/4.0/。转载和许可
引用这篇文章

Kovalchik, K.A., Hamelin, D.J., Kubiniok, P.
等人。Machine learning-enhanced immunopeptidomics applied to T-cell epitope discovery for COVID-19 vaccines.纳特·康姆15 , 10316 (2024). https://doi.org/10.1038/s41467-024-54734-9下载引文
:2024 年 1 月 31 日
:2024 年 11 月 20 日
:2024 年 11 月 28 日
:https://doi.org/10.1038/s41467-024-54734-9https://doi.org/10.1038/s41467-024-54734-9