
使用无监督深度学习综合增强癌细胞系多组学数据集
作者:Gonçalves, Emanuel
介绍
癌细胞系不断增长的分子和表型特征使其成为研究最多的人类细胞模型之一1。这种不断增长和丰富的多组学数据继续推动癌症基因的识别和治疗靶点的发现2,3,4。尽管基因组学一直是寻找癌症预测生物标志物的主要焦点,但癌症依赖性图谱 (DepMap) 联盟最近进行的功能遗传筛查显示,不到 20% 的 RNAi 癌症依赖性可以通过突变和拷贝数改变来解释5。这凸显了开发能够垂直集成正交数据集的整体机器学习模型的重要性。在这种情况下,垂直整合不仅涉及基因组学,还涉及其他类型的组学数据6。
尽管深度学习最近取得了成功7多组学整合面临着一些限制,最重要的是不同数据类型的高度异质性(例如,离散分布与连续分布)、固有的技术限制(例如,缺失值)和有限的数据可用性(例如,在本研究中,只有 25.8%)的癌细胞系拥有一整套正在考虑的所有七个组学数据集)8。无监督机器学习在多组学集成中取得了成功,捕获了不同组学之间共享的数据变化模式9,10。这种方法强调了与上皮间质转化(EMT)相关的癌细胞状态,这是耐药性和转移的关键过程11。基于无监督深度学习的模型可以通过重建缺失的测量值和纠正实验误差来生成输入数据集的改进版本,从而增强下游分析12,13。虽然线性降维模型10,14虽然深度生成模型的设计目的相似,但其在大规模多组学癌细胞模型中的应用却相对滞后。这就在利用这些非线性方法来扩充数据集和进行统计分析以改善癌症机制、生物标志物和药物靶标的表征方面留下了空白5,15,16。深度学习模型,例如变分自动编码器(VAE),提供了更复杂的基础生物数据公式。此外,VAE 具有高度灵活的设计,可以稳健地处理数据稀疏性,并且可以轻松扩展以合并不同的数据类型。特别是,基于 VAE 模型的方法在单细胞多组学整合和增强领域取得了巨大成功。然而,这些方法通常预设特定数据类型的存在,例如来自 scRNA-seq 和 scATAC-seq 的计数数据,限制了它们在更广泛的组学领域的适用性17 号,18,19,20,21。
在这里,我们开发了一种多组学综合增强 (MOSA) VAE 模型,该模型集成并综合增强了来自 DepMap 超过 1500 个癌细胞系的多组学数据集。MOSA 提供了一种用于癌症发现的生成式无监督深度学习模型,该模型利用 SHapley Additive exPlanations (SHAP)22模型可解释性的价值,有助于识别潜在的生物机制和药物靶点。在我们的研究中,我们系统地评估了 MOSA 并对其进行了基准测试,证明了其在独立药物反应和蛋白质组数据集上的生成能力,以及准确恢复癌症组织起源聚类的能力。此外,MOSA 还提高了通过 CRISPR-Cas9 基因必要性筛选发现基因组关联的统计能力。综合筛选的癌细胞系揭示了与基因组图谱一致的漏洞,例如弗利1-EWSR1融合依赖性。借助 MOSA,我们生成了涵盖所有七个不同组学的完整多组学概况,使可用屏幕数量增加了 32.7%。
结果
统一癌症多组学的深度生成模型
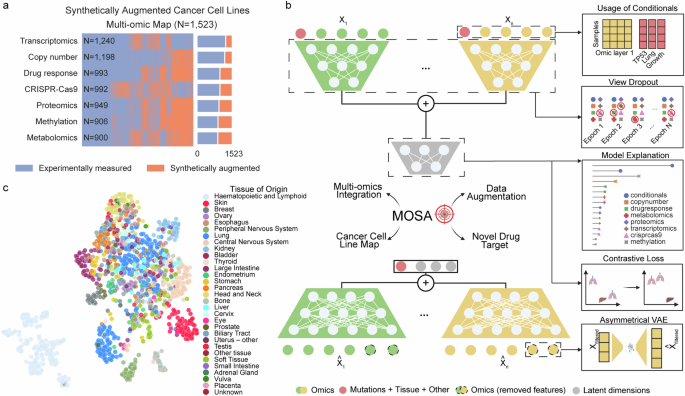
利用 DepMap 项目5,6,23,24,我们组装了七个不同的癌细胞系数据集,即基因组学2,3, 甲基组学25, 转录组学26, 蛋白质组学11, 代谢组学27, 药物反应2,25,28,29和 CRISPR-Cas9 基因必要性4,30(图. 1a)。这总共包括 1523 个癌细胞系,其中至少有两个数据集可用(补充数据 1)。我们设计了针对癌细胞系多组学数据集的 MOSA,进行了稳健的数据增强,并为生物标志物的发现提供了模型解释(图 1)。 1b,参见方法)。图 1:癌症多组学与 MOSA 的整合。

涵盖 1523 个癌细胞系的癌细胞系多组学数据集。紫色代表测量的屏幕,而橙色代表间隙,即缺失的屏幕,这是用 MOSA 合成生成的。乙自动编码器 MOSA 的示意图,其中编码器表示在顶部,解码器表示在底部。为简单起见,仅表示两个数据集的集成。MOSA 的突出设计如右图所示。在 BioRender 中创建。蔡正 (2023)BioRender.com/m96b457。c使用训练后的 MOSA 关节潜在空间的统一流形逼近和投影 (UMAP) 表示进行可视化降维,其中每个点代表根据其来源组织着色的癌细胞系。首先,在后期整合之后31
方法中,我们为每个数据集训练了一个单独的编码器,以导出特定于每个组学层的潜在嵌入。然后将这些嵌入连接起来并进一步简化以形成联合多组学潜在表示(图 1)。 1c, 补充数据 2)。这里,潜在表示是神经网络隐藏层中学习的、抽象的特征集(嵌入),它封装了输入数据中的主要信息。与多组学线性降维方法相比,MOFA10,14,以及另一种基于 VAE 的方法 MOVE32,我们的模型可以在多组学潜在空间中通过组织更好地分离细胞系(图 1) 1c,补充图。 1)。
其次,由于数据的稀疏性和定性性质,基因组学提出了独特的挑战。为了解决这个问题,我们仅使用癌症驱动事件,并将基因组学分为拷贝数改变和突变。虽然拷贝数事件通过类似于其他组学的单独编码器/解码器集成为序数数据,但突变作为二进制条件集成到每个编码器(图 1)。 1b,参见方法)。其基本原理是遗传背景影响细胞特征和表型,从而调节其他组学层。条件矩阵包含癌症驱动基因的遗传改变(包括基因融合)、细胞系组织来源、细胞系生长率测量值和微卫星不稳定性信息(MSI 高),总共 237 个条件变量(补充数据) 3)。该条件矩阵进一步连接到学习的多组学联合潜在空间,作为解码器的输入。因此,遗传背景和细胞信息对于生成潜在表示和重建每个组学数据集至关重要。
三、与单细胞数据的同类模型比较12,21,33,DepMap 中可用的样本数量有限和组学异质性对训练癌细胞系的通用模型提出了重大挑战。为了降低模型复杂性,MOSA 只考虑最可变的特征作为编码器的输入,而所有特征都由解码器重建以生成合成数据,从而导致 VAE 的不对称设计(图 1)。 1b,补充图。 2a、b, 补充数据 4)。MOSA 的这种独特设计使我们能够丢弃信息量低的特征,例如所有癌细胞系中恒定表达的基因和非必需基因。这将可训练参数的数量减少了 39.2%,同时保持了较低的重建误差。
第四,组学多组学数据集的不同大小可能会导致某些数据集在训练过程中占主导地位,从而降低模型的通用性和可解释性。我们开发了一个完整的组学(视图)dropout 层,它基于超参数掩盖了完整的组学层。这显着改进了模型的泛化能力,通过特定组学为癌细胞系提供了更好的重建(参见方法,图 1)。 1b)。然后,我们通过计算 SHAP 值来执行多组学模型解释22对于所有组学输入特征,以评估它们对于潜在空间整合和组学特征重建的重要性(参见方法)。这为探索潜在的非线性癌症基因型-表型关联提供了系统资源。
总而言之,MOSA 提供了一个同时整合所有癌细胞系组学的无监督模型。使用 10 倍交叉验证策略,MOSA 重建的 CRISPR-Cas9 保留折叠和药物反应与原始数据密切相关(平均特征 Pearson s r 分别为 0.35 和 0.65)(图 1)。一个 2a,补充图。 3, 补充数据 5)。与类似的系统监督分析相比,MOSA 的表现更好,该分析旨在使用核心组学(例如基因组学、转录组学)、仅基因组学或仅功能相关基因(平均特征最佳 Pearson s r)来预测每个 CRISPR-Cas9 基因依赖性。=0.25)34。

MOSA 重建质量使用 10 倍交叉验证进行测量。重建所有测试折叠后,将它们连接起来,并将重建质量分数计算为重建值和实际测量值之间的 Pearson r。显示了药物反应(左)和 CRISPR-Cas9(右)数据集的按重建质量排名的特征。重复的药物名称代表同一药物的重复筛选。强选择性 CRISPR-Cas9 和药物反应的代表性例子已被标记。乙与最近的独立药物反应筛选相比,MOSA 的药物 IC50 的部分数据集增强(缺失值插补)。c—e,类似于乙,分别使用 MOFA、MOVE 和平均估算值。多组学合成数据生成的评估
多组学垂直整合和无监督深度生成模型的一个显着优势是它们能够综合生成特定样本中缺失的数据集,例如重建某些细胞系完全不存在的数据集。
考虑到普遍存在的数据集差距,这一点尤其重要,即使是在癌细胞系等特征良好的模型中(图 1) 1a)。多组学分析既昂贵又费力,因此数据驱动的生成模型是优先设计信息最丰富的实验的关键。然而,对生成模型进行基准测试具有挑战性,因为它需要独立的、最好是大规模的数据集来验证模型的预测。我们最初测试了 16 种多组学整合方法(补充数据 6,参见方法),但由于受支持的组学数量、数据分布类型以及设计和实施方面的限制等限制,我们将范围缩小到三种最先进的方法: MOFA10,14, 移动32和混合组学35,36。这些方法包括线性、基于 VAE 和相关分析方法,能够整合此处考虑的所有七个组学数据集。我们在以下部分中描述了一系列提高模型复杂性的基准。
MOSA 利用从原始数据中学习到的多组学潜在空间来重建输入数据矩阵。数据重建生成完整的组学矩阵,从而处理缺失值(部分数据集增强),更重要的是,通过垂直整合重建整个组学(完整数据集增强)(在此考虑的细胞系至少需要两个组学)学习)。对于部分数据集扩充,MOSA 会估算不完整的特征,例如,由于基于质谱的蛋白质组数据中常见的技术限制,某些蛋白质的测量结果稀疏11,37。准确地重建了模型训练期间完全不存在的最新独立药物反应数据集(IC50,Pearson r = 0.87,n≤=≤32,659)(图 2b),优于 MOFA10,14, 移动32和朴素平均插补(图。 2câe)。MOSA 的重建和原始数据集之间的明显差异揭示了实验测量可能不准确。例如,对 MEK1/2 抑制剂曲美替尼 (Trametinib) 的反应与重复测量和同一细胞系中具有相同规范靶点的药物不一致(补充图 1)。 4a)。这种差异还突出了没有有效分子生物标志物的药物(例如维奈托克)或药物类别(例如抗凋亡抑制剂)(补充图1) 4b),强调了为他们的反应设计可靠的预测模型的挑战。此外,蛋白质组学充满了缺失值(补充图 1) 5a),主要影响低丰度蛋白质。MOSA 通过使用来自所有组学的信息填充约 32% 的原始矩阵来增强蛋白质组数据,同时保留样本与独立蛋白质组数据集 (CCLE38)(补充图。 5b, 补充数据 7)。值得注意的是,MOSA 有效地重建了SMAD4在具有以下特征的细胞系中SMAD4基因缺失,通常与低水平相关SMAD4基因表达和蛋白质丰度(补充图。 5c)。MOSA 增强的蛋白质组矩阵保留了通过蛋白质成对相关性识别蛋白质相互作用的能力39(补充图 5天)。与具有缺失值的原始矩阵相比,MOSA 的增强蛋白质矩阵是完整的,可直接用于下游分析,例如广义线性模型40,这提高了蛋白质复合物相互作用的回忆(补充图 1) 5天)。
随后,评估了完整的数据集增强。MOSA 为缺乏蛋白质组测量的癌细胞系生成的合成蛋白质组数据显示出与独立蛋白质组测量的相关性,与具有实际蛋白质组数据的细胞系的数据相当(图 1)。 3a)。对于药物反应,107 种重叠药物的重建与独立数据集 (CTD241,42)(图。 3b)。最后,我们使用独立处理的转录组学进行了类似的分析,其中包括在 MOSA 训练期间没有转录组学数据的 272 个癌细胞系的数据26。MOSA 的转录组重建与真实数据密切相关,即使对于没有转录组数据进行训练的细胞系也是如此(平均 pearson s r = 0.90)(补充图 1) 5e)。至关重要的是,这表明 MOSA 作为合成癌细胞系多组学和表型筛选的生成模型的能力。

蛋白质组学癌细胞系分布与独立数据集的相关性(CCLE38)根据癌细胞系是否具有用于模型训练的蛋白质组数据进行分组(橙色,n–= –291) 与之前没有任何蛋白质组学的细胞系(浅蓝色,n≤=≤78)。乙独立药物反应数据集 (CTD2) 之间癌细胞系相关性 (Pearson-s r) 的分布41,42)和 MOSA 重建数据集,根据模型训练数据集中癌细胞系是否具有先前的药物反应可用性进行分组(橙色、n–= –571) 与没有药物反应数据的细胞系(浅蓝色,n–= –239)。c原始数据集(x 轴)和增强 MOSA 数据集(y 轴)与 CRISPR-Cas9 基因必要性的遗传关联的单侧对数比测试 p 值。使用 Benjamini-Hochberg 方法应用错误发现率 (FDR) 校正来调整多重比较。d在原始 CRISPR-Cas9 数据集(x 轴)和 MOSA 增强数据集(y 轴)上对每个基因进行 Fisher 偏斜测试。点大小表示原始数据集中具有必需基因的细胞系数量(缩放 log2 倍数变化≤<≤0.5)。e之间的相关性布拉夫和MAPK1使用先前测量(观察)和综合重建(重建)的 CRISPR-Cas9 基因必需性。基因重要性分数用校正后的拷贝数表示78log2 倍数变化按常见必需基因(得分≤=≤1)和非必需基因(得分≤=≤0)的中位数缩放30。基因必要性也根据存在或不存在进行分组布拉夫突变,主要是V600E功能获得突变。fCRISPR-Cas9 基因重要性与弗利1-EWSR1融合。面板中的回归线显示 95% 的置信区间d,e, 和f。盒须图显示 1.5 × 四分位数范围,中心表示面板中的中位数e和f。我们通过将原始数据矩阵与增强数据矩阵进行比较来评估下游分析。MOSA 将 CRISPR-Cas9 细胞系筛选的数量增加了 34.9%,增强的数据集提高了寻找遗传关联的统计能力(图 1)。
3c, 补充数据 8)。基因必要性特异性(Fisher 偏度检验)可用于识别选择性癌症脆弱性,在合成的 CRISPR-Cas9 筛选细胞系之间显示出中度正相关性 (Pearson r = 0.52)以及之前可用的屏幕(图 3d)。尽管如此,由于潜在异常非必需基因的存在,这种相关性可能被低估。MOSA 准确地重建了基因依赖关系,例如,布拉夫依赖于布拉夫功能获得性突变癌细胞系(图。 3e), 和弗利1细胞系的依赖性弗利1-EWSR1融合基因(图1) 3f)。
最后,我们的目的是评估开发一种能够本地整合两个以上组学的方法的优势。具体来说,我们专注于转录组学和药物反应数据集,它们分别代表分子和表型数据集。这些也常用于多组学集成,并且是我们基准测试中信息最丰富的组学类型之一。从我们评估的方法列表中,我们考虑了 iClusterPlus43, 杰米18,scVAEIT44和 moCluster45(补充数据 6,参见方法)。MOSA 提供了更好的转录组学和药物反应数据重建(补充图 1) 6a、b)。特别是,在 MOSA 中添加更多组学比现有方法有了显着改进,支持使用整体多组学模型的实用性。此外,MOSA 在组织起源聚类方面始终优于其他方法(补充图 1) 6c)。仅考虑转录组学和药物反应会产生最佳的起源组织聚类,反映了这些组学按起源组织的强大结构46。相比之下,蛋白质组学和代谢组学等其他组学的组织结构更为松散11,47。因此,包括组织结构不太牢固的组学自然会导致组织聚类更加松散。
总而言之,这些不同的例子证明了 MOSA 执行部分和完整数据集增强的能力,并使用来自不同实验室的各种独立数据集进行了验证。大规模多组学数据集的生成既耗费时间又耗费资源,因此 MOSA 成为计算机测试和药物靶点优先排序以进行实验验证的宝贵工具。
模型解释揭示癌细胞状态
为了优先考虑最有希望的目标,模型除了产生可靠的预测之外还需要具有可解释性。因此,我们使用了SHAP22计算特征重要性的算法,定义为每个特征对潜在空间的贡献量(图 1) 1b, 补充数据 9,参见方法)。当按相应的组学数据集对特征进行分组时,我们观察到代谢组学、药物反应和拷贝数改变表现出最高的平均特征重要性(补充图 1)。 7a)。关于条件特征,虽然它们的平均特征重要性不大,但某些关键特征,例如TP53即使与其他组学数据集相比,突变、生长率以及造血和淋巴起源的组织也显得非常显着(图 1)。 4a, 补充数据 9)。这强调了将条件变量纳入模型的重要性。每个组学数据集中排名前五的特征也验证了我们的方法恢复与癌症相关的成熟分子过程的能力(图 1)。 4a), 例如,CDKN2A拷贝数改变以及对 SRC 家族抑制剂达沙替尼的敏感性。有趣的是,其他排名较高的不太明显的特征揭示了以前较少探索的生物机制。一个具体的例子是参与烟酸盐和烟酰胺代谢的代谢物 1-甲基烟酰胺,它被认为是多组学潜在表征的代谢组学中最重要的特征(图 1)。 4a)。我们观察到细胞内 1-甲基烟酰胺丰度的增加与烟酰胺 N-甲基转移酶 (NNMT) 的过度表达之间存在密切关系,该酶催化这种代谢物的产生(补充图 1)。 7b)。我们还观察到 1-甲基烟酰胺与癌细胞系 EMT 状态之间的关联,这一点得到了维姆和CDH111(补充图 7c、d)。这证实了最近的一项单细胞研究发现,PC-9 非小细胞肺癌细胞系具有激活表皮生长因子受体突变,通过 EMT 标记物的表达以及 1-甲基烟酰胺的积累形成对 EGFR 抑制剂耐药的细胞状态48。此外,在 A549 细胞系的 EMT 早期阶段观察到 1-甲基烟酰胺显着增加,这种增加与糖酵解代谢物和组蛋白翻译后修饰的变化有关,表明 1-甲基烟酰胺与表观遗传修饰之间存在联系急救期间49。虽然需要进一步的实验验证,但这可能为识别耐药性背后的癌细胞状态铺平道路。图 4:MOSA 的 SHapley Additive exPlanations (SHAP) 模型解释。一个

乙代谢物 1-甲基烟酰胺具有最高特征重要性的顶级药物。c对达拉普林(乙胺嘧啶)药物反应重建贡献最大的主要特征。为了更深入地研究,我们随后使用 SHAP 算法来计算专门用于重建药物反应的特征重要性,从而促进发现最有希望的生物标志物(参见方法)。正如预期的那样,平均而言,药物反应特征本身是最重要的特征(补充图 1)。 8a
, 补充数据 10)。值得注意的是,条件成为第二重要的组学,反映了组织的起源、突变和生长速率在影响药物反应中的关键作用(补充图1)。 8a)。以代谢物 1-甲基烟酰胺为中心,已知与 EMT 相关的药物被列为显示 1-甲基烟酰胺高度特征重要性的顶级药物(图 1)。 4b)。最近的研究发现,除达拉匹林(乙胺嘧啶)未作为抗癌药物纳入数据集中外,排名前五的药物均与 EMT 相关。具体来说,UNC063850, 恩替司他51、BIX0218952甲氨蝶呤抑制 EMT53显示出诱导 EMT 的能力。这一发现表明排名第一的药物 Daraprim 也可能与 EMT 有着密切的关系,为癌症治疗的重新利用提供了潜在的途径。其他 EMT 相关特征,例如 GPX1 蛋白强度54也被列为 Daraprim 的首要特征,表明有可能利用列表中的其他特征来发现最有希望的药物反应生物标志物(图 1)。 4c)。在顶级药物的其他主要功能中,克拉斯和KMT2D一直被认为具有高度重要性,并且这两个基因都与 EMT 有关55,56(补充图 8 点)。最后,我们利用了外部代谢组数据集47通过 SHAP 值验证与 1-甲基烟酰胺相关的药物。尽管在他们的研究中没有直接测量 1-甲基烟酰胺的丰度,但我们分析了与烟酸盐和烟酰胺代谢相关的药物,其中 1-甲基烟酰胺是烟酰胺甲基化的直接产物。由 SHAP 值识别的几种突出药物,包括 Daraprim、UNC0638、Entinostat (MS-275) 和 PAC-1,也被列为耐药或敏感药物(补充图 1)。 9)。
综上所述,我们的研究结果表明,1-甲基烟酰胺和 EMT 在数百种癌细胞系中存在广泛关联,并在耐药性中发挥潜在作用。虽然需要进一步评估来证实这一点,但更广泛地说,它揭示了使用 MOSA 作为整体模型的可能性,该模型整合癌细胞的分子和表型数据来研究癌细胞状态、耐药性及其潜在机制。
讨论
包括 MOSA 在内的深度生成模型在癌症研究中的应用前景广阔,但也存在局限性,主要与样本量有限有关,这阻碍了探索更复杂的 VAE 设计,而更复杂的设计导致数据集重建更差。虽然数据集的整体重建是稳健的,但也有一些可以改进的例子,特别是对于蛋白质组学,其中固有的数据稀疏使得模型成功训练更具挑战性。因此,添加更具特征的癌症模型可能会让我们训练更好的模型并减少重建误差。未来的努力应该利用来自癌症患者和衍生模型(例如类器官和患者衍生异种移植物(PDX))的多组学资源,以加强培训并探索转移学习机会。除了表格组学数据之外,VAE 在集成图像和基于文本的数据方面也取得了巨大成功57,58,并且 MOSA 可以进一步增强以集成这些类型的数据并实现多模式数据增强。我们还旨在通过采用 VAE 架构来更准确地识别和处理 MNAR 场景,解决数据非随机丢失 (MNAR) 的复杂挑战,这是组学数据集中常见的场景59,60,61。SHAP分析为深度学习模型提供了解释,然而,在验证某些突出特征的生物学意义方面仍然存在一些障碍。这些挑战可能与 SHAP 和 Shapley 值的固有局限性有关62,因此需要进行额外的研究来确定这些强调特征的重要性。此外,虽然这为 EMT 相关关联提供了强有力的初步支持,但还需要进一步的实验工作来在不同的癌细胞模型中验证和确认这些发现。
总之,MOSA 通过有力地填补现有实验筛选中的空白,增强了 1523 种癌细胞系的多组学特征。基于深度学习的合成数据生成可以通过促进真实数据集的创建来增强实验屏幕,以指导实验设计并加速最有希望的目标的验证。展望未来,该模型很容易适应集成其他类型的数据模式,例如成像,进一步促进分子/表型关联的发现。
方法
癌细胞系多组学数据收集
目的是组装最新,全面的分子,表型和癌细胞系样本信息。所有数据集从DepMap下载(https://depmap.org/),以及CellmodelPassports(https://cellmodelpassports.sanger.ac.uk/)23门户,除代谢组学数据外,这些数据直接从原始出版物补充材料中获取27。为了获得可重复性,本研究中使用的所有数据均在Figshare存储库中提供(请参见代码和数据可用性)。
我们整合了基因组学2,63,转录组学26,甲基组学25,蛋白质组学11,代谢组学27,药物反应25,28,29和CRISPR-Cas9基因的必要性4,64。这总共包括1523个癌细胞系,至少有两个数据集可用于每条细胞系。所有数据集先前均已处理,标准化/缩放和批处理,并在其每个个人出版物中都纠正了针对每个数据集很重要的技术和设计方面(例如,在不同实验室的CRISPR-CAS9屏幕集成65,驱动程序突变和拷贝数更改以及来自不同数据集的基因表达样本26)。
癌细胞系验证数据集
本研究中使用了三个独立的数据集进行验证,即未用于模型培训。第一个数据集介绍了375种癌细胞系的CCLE蛋白质组学表征38,其中291个包含蛋白质组学数据集11用于训练。第二个数据集代表与用于培训的药物筛查相同平台的最新药物反应筛选25,28,29是从癌症(GDSC)门户中药物敏感性的基因组获得的(https://www.cancerrxgene.org/)66总共包括32,659个IC50,在313种独特的药物和781个重叠的癌细胞系中测量。第三个数据集是独立的药物响应数据集(CTD2)41,42,总共包括545种药物和887个癌细胞系,分别为106和575,与用于训练的药物反应数据重叠25,28,29。
数据预处理
总共考虑了七个数据集:复制号(n= 777个功能);甲基n= 14,608);转录组(n= 15,278);蛋白质组(n= 4922);代谢组(n= 225);药物反应(n= 810);和crispr-cas9基因的必要性(n= 17,931)。总共1523个癌细胞系的每个细胞系具有至少两个数据集。
对于CRISPR-CAS9基因的必要性,进行了转录组和甲基甲基分子特征的降低以排除较低的可变特征。对于基因的本质,分别使用必需基因和非必需的基因将样品缩放为每个样品-1和0的中位数。从来没有丢弃基本基因,即没有基本特征的基因低于至少一个细胞系中必不可少基因的中值log2折叠变化的50%。对于转录组学和甲基组学,应用了标准偏差过滤器。通过将所有基因的标准偏差跨样品进行,拟合了高斯混合模型(Kâ= 2),从而识别降低的基因和其余基因。标准偏差阈值定义为两个高斯分布的最右截距(补充图。 2a),以及标准偏差低于丢弃的任何基因。此外,对于蛋白质组学,药物反应,代谢组和CRISPR-CAS9数据集,丢弃了率高于85%的任何功能。除复制号外,所有数据集均通过Z分数标准化。缺少值被0替换为0,它们在原始数据集中的位置存储在模型中(例如,将它们排除在损失函数之外)。除这七个数据集外,还将驱动基因突变,融合基因,微卫星不稳定性,生长速率,癌症和组织类型信息连接到单个矩阵中,以用作癌细胞系的标签。
多词合成增强(MOSA)
MOSA是使用Pytorch(v2.0)实现的条件多视图变异自动编码器67。在下一部分中,我们描述了MOSA的架构,条件使用,辍学层和塑造性分析。
建筑学
MOSA遵循传统的条件VAE设计(图 1b)。对于七个数据集(视图),编码器均经过多个完全连接的层进行训练,这些图层与数据集的输入功能的数量以及标签数(串联条件)成正比。首先,关节完全连接的层将每个数据集输入,并将其减少到固定数量的关节潜在尺寸。测试了不同的技术,以整合奥马特异性的潜在维度(例如专家的乘积),但串联获得了最小的重建损失。多矩联合潜在维度进一步降低为指定数量的潜在维度(超参数)。然后,关节层输出两个代表高斯分布平均值和方差的层。这些对于潜在空间的正则化很重要,用于采样潜在的维度。最后,将潜在尺寸(Z)与条件串联并提供给每个数据集的解码器。解码器具有与编码器相似但反相反的架构。
条件句
我们引入了条件体系结构,以增强模型的重建性能和生物学相关性。有条件(n237)包括关键的生物学特征,例如癌症驱动突变,组织类型,基因融合,MSI状态和细胞系生长速率。这些在模型体系结构中分为两个阶段:1)在编码之前将其连接到每个Omic层;2)在解码之前与多摩尼克联合潜在表示。条件串联具有两个至关重要的目的:它在特定的细胞或遗传背景中将输入数据进行了背景,并且允许解码器生成数据的特定条件重建。包含有条件的有几个优势。首先,它确保该模型不仅是孤立地捕获单个Omic层中的模式。取而代之的是,考虑到多摩尼克数据之间的复杂相互作用以及基因组和生理变量,促进了对基本生物学过程和现象的更全面的理解。其次,通过将这些条件嵌入解码器中,该模型可以生成与特定细胞线条件相关的数据重建。
查看辍学层
MOSA中包含了一种特殊的辍学策略,即视图辍学层,以提高模型的预测能力和解释性。与传统的辍学层(随机将单个特征都设置为零)不同,视图辍学层零将单个OMIC层的所有输入特征零。这种方法鼓励模型通过学习多个OMIC层之间的关系,而不是依靠一个特定的OMIC层来重建数据。例如,在产生药物反应预测时,MOSA模型可能不成比例地强调输入药物反应数据,从而忽略了其他OMIC的潜在贡献,例如转录组和蛋白质组学数据。通过使用视图辍学层,我们显着改善了潜在的空间细胞系分离(图 1c,补充图 1b,10a,b)和重建蛋白质组学(图 3a,补充图 10c)和药物反应数据(补充图。 10天)。该层的辍学率由超参数View_dropout控制,最终模型的最佳设置为0.5。
通过Shapley添加说明(SHAP)的模型说明
对于模型说明,我们使用了Python包装的形状22(v0.42.1)具有技术修改,以支持多摩变数据作为MOSA的输入。具体而言,结合了集成级的梯度解释器68和Smoothgrad69,用于计算模型输出上有关输入的梯度的变化,以将重要性值归因于每个功能。形状计算以两种方式进行。
首先,运行Shap来解释MOSA的编码部分,将综合的潜在维度视为输出。结果包含在多维阵列中的形状值\((({n} _ {{litent \ _} \ dim},\,{n} _ {{samples}},\,\,{n}} _ {{n}} _ {{features {features}})\),其中每个\(n \)分别表示潜在维度,样本和特征的数量。为了达到分析的全球水平特征的重要性,首先将多维阵列作为解释正面和负面影响的绝对值,然后在潜在维度上求和,然后通过样本进行平均。然后这导致了长度列表\({n} _ {{features}} \),表示有助于潜在空间的整体特征重要性(图 4a)。
其次,运行Shap来解释MOSA的每个OMIC数据集的重建。以药物反应为例,类似于解释潜在空间,形状值的形状为\(({{n} _ {{drugs}},\,{n} _ {{samples}}},\,{{n}}} _ {{features}})\)\)\), 在哪里\({n} _ {{drugs}} \)\)代表毒品的数量,并且\({n} _ {{samples}},\,{n} _ {{features}}} \)被描述为上述。在此分析中,阵列仅在样品中平均,导致2D阵列\((({n} _ {{drugs}}},\,{n} _ {{features}})\)\),衡量对每种药物的特征重要性。首先选择了特征1-甲基二酰胺代谢产物,并分析具有最高外形值的药物以鉴定与EMT相关的药物(图。 4b)。然后,通过选择药物行,然后在降序中对特征进行排名,对感兴趣药物的其他重要特征进行排名(图 4c)。由于计算资源的限制,随机选择了20%的样本以计算重建药物响应和拷贝数数据集的特征重要性,而将20个样品随机选择用于其他具有较大尺寸的OMIC数据集。
总体而言,外观分析使我们能够确定对多摩尼克潜在维度很重要的特征,并解释了特征的重建,例如药物反应。在所有样本中汇总的特征和输出尺寸汇总的特征可以在补充数据中找到 9和10。对于每个输出维度的更重要的特征可以从中下载代码和数据可用性部分。
损失函数
损耗函数是三个组成部分的总和:1)所有输入数据集的重建误差;2)多运动联合维度的加权变异kullbackâleibler(KL)正则化项70;3)使用组织类型作为标签的对比损失:
$$ {{{损失}} _ {{attry}} \,= \,{l} _ {{reconstruction}}}+\ lambda {l} _ {{kl}}}}}$$
(1)
重建损失\(\,{l} _ {{reconstruction}} \)定义为:
$$ {l} _ {{reconstruction}} = {\ sum} _ {d} {l} {l} _ {d} $$
(2)
在哪里\({l} _ {d} \)表示数据集的重建损失d,使用平均误差(MSE)计算71,72。在等式中。(
1) 这我”并且分别是优化的超参数,以分别加重KL差异和对比度损失项。\({l} _ {{kl}} \)计算有平均值的学识渊博的高斯分布之间的KL差异(\(\亩\))和差异(\({\ sigma}^{2} \)VAE和标准的正常先验分布70。损失函数的最后一部分是对比损失,定义为:
$$ {l} _ {{contrastive}} = [{m} _ {{pos}}} - {s} _ {p}] _ {+}+[{s} _ {s} _ {n} - {m} - {m} _ {m} _ {
{neg}}] _ {+} $$
(3)
在哪里\({s} _ {p} \)和\({s} _ {n} \)表示正对和负对之间的余弦相似性,这是由两个样品是否具有相同的组织类型来定义的。\({m} _ {{pos}} \)和\({m} _ {{neg}} \)是正缘和负边缘,它们是按照以下部分所述调整的超参数。
不对称的vae
MOSA还采用不对称结构设计,以减少参数数量来优化模型效率。具体而言,在编码过程之前以数据类型的方式进行了特征选择。对于转录组和甲基化数据,仅选择表现出较高可变性的特征作为模型的输入。使用高斯混合模型定义了高度可变的特征,其两个组件拟合到所有功能的标准偏差,从而捕获了低较低且高度可变的特征的两个分布。标准偏差阈值定义为两个分布的密度相等的最大值,因此,转录组数据的标准偏差大于1.122,甲基化数据的特征被认为是高度可变的,被认为是编码器的输入。对于CRISPR-CAS9数据,从输入层中排除了基因适应性得分高于每个细胞系的基因适应性评分高于0.5的基因敲除,这是基因适应性得分高于0.5的基因敲除。这种目标特征选择有效地减轻了模型的计算负担。尽管输入复杂性降低了,但在解码过程中包含所有可用功能以重建数据。选择这种不对称的设计是因为其能够在简化其体系结构的同时保持模型的预测和重建能力的能力。
超参数
选择超参数(表 1)以基于平行试验的自动优化框架为指导(Optuna73),然后手动调整。对于每次运行,进行分层的洗牌分裂,通过造血和淋巴样细胞系进行分层,留下20%的样品进行测试。总共进行了600次试验,每个试验都限制为150个时期。
基准的最新方法
为了与MOSA采用的无监督的多词方法进行比较,测试了16种多摩尼斯整合方法。但是,如补充数据中所述 6,对于大多数模型,我们遇到了与内在设计和实现选择有关的问题。这些包括对支持的OMIC模式的数量和仅针对计数数据处理量身定制的特定设计的限制,因为许多模型都是为单细胞数据设计的。因此,我们设法对所有七个OMIC数据集进行了运行和系统的基准测试我们的结果,该数据集针对其他三种用于多摩变数据集成的最先进方法,包括MOFA10,14作为降低线性多摩尼克的线性方法,移动32作为基于VAE的方法和混合学35,36这是基于广义规范相关分析。为了使比较尽可能接近并仅关注方法论上的差异,将相同的数据预处理用于MOSA,MOFA,MOVE和MIXOMICS。同样,最初将MOFA的因子数量设置为相同最佳的联合潜在维度,即200(表格) 1)。但是,这产生了性能不佳的结果,即重建的数据集很差。通过手动探索,最佳因素数量设置为100,这在训练期间自动降低至97,通过丢弃差异的因素低于0.0001。每个视图都独立缩放,并运行该模型直到收敛(Contgence_Modeâ=慢)。尺寸的数量成功地将其设置为200。由于混合学需要与每个OMIC数据集分别关联的维数的数量,因此在混合学中使用了210个维度来实现最接近的比较。MOSA中的条件层均包含二元,例如原产的突变和组织,以及连续的,例如,癌细胞系的生长速率和独立研究的倍增时间,特征。这些数据作为移动中的单独层包括在内,但是,MOFA不支持混合的分布式视图,例如高斯和伯努利。因此,在条件视图中,我们无法整合增长率和加倍时间,除了这两个视图之外,它们具有二进制特征,因此将先前的可能性分布设置为伯诺利。这些配置产生了使用MOFA的最佳多摩变维度降低并查看重建。优化的模型被保存为HDF5文件,并在Figshare存储库中提供。类似地,在包装文档后,将原始数据的组织数据用作混合组学的目标变量。具体而言,暗黑破坏神模式36(N-整合)在Mixomics Suite中,这是原始Mixomics Toolset的扩展35在本研究中,用于多摩斯数据集成任务。
为了更全面地评估MOSA,并解决了仅支持有限数量的OMIC模式的许多最新方法的局限性,我们进行了单独的基准分析分析,仅考虑两种OMIC模式为输入。这种方法允许我们包括四种其他方法:杰米18,scvaeit44,iClusterPlus74和mocluster45。转录组和药物反应数据用于针对其他七种方法训练和基准MOSA,因为这些OMIC类型是我们基准的重点。由于仅包括两个OMIC,因此我们删除了对样本至少有两个OMIC的数据的要求。Jamie,Iclusterplus和Mocluster使用200个维度成功地运行了与MOSA相同的集成潜在空间。但是,具有200个潜在维度的SCVAEIT产生的结果差,尤其是对于潜在的空间聚类比较。与MOFA类似,我们手动搜索了最佳设置,并决定使用100个潜在维度进行SCVAEIT。此外,对于转录组和药物反应数据的Dist_block超参数设置了高斯分布。评估了MOSA,MOFA,MOVE,JAMIE和SCVAEIT的合成数据重建和聚类性能,而Mixomics,IclusterPlus和Mocluster仅包括在潜在的空间群集比较中,因为它们不是生成模型。为了确保公平的比较,在训练过程中未将条件纳入任何选定的方法中。
蛋白质 - 蛋白质相互作用共同扩展分析
使用两种方法比较MOSA增强蛋白质组学基质的能力和原始蛋白质组学矩阵来概括特定蛋白质相互作用资源中存在的PPI的PPI:CORUM75,Biogrid76和字符串77。第一种方法是Pearson的S R,以前已用于此任务39。由于其固有的局限性,例如,它无法说明混杂效应和数据结构,这是一种新方法,类似于Wainberg等人所描述的方法。40测试了基于广义线性模型(GLM)。该方法通过使用其协方差矩阵将cholesky的美白转化应用于蛋白质组学数据,该矩阵将样本解释并将数据推向正态性。然后将此转换的数据用于普通最小二乘(OLS),其计算的权重是两个蛋白质之间的相关度量。计算每种蛋白质对的每种方法,通过上升p值对每种方法排序所有对。之后,根据PPI集中的该对的累积总和来绘制曲线,\(\ frac {1} {k} \)(存在)或0(不存在),其中\(k \)是当前对的总数。因此,方法越好,回忆曲线的AUC越大。
统计和再现性
样本量是通过癌细胞系的可用性和癌症依赖图(DEPMAP)的相关多OMIC数据集确定的。总共包括1523个癌细胞系,至少有两个数据集可用。没有使用统计方法来预先确定样本量。数据排除细节可以在方法中的数据预处理部分下找到。数据被随机拆分以进行交叉验证。通过造血和淋巴样细胞系对随机进行分层,以确保跨细胞系类型和不同培养条件的平衡表示,即悬浮液与粘附。盲目不适用于本研究,因为所有分析均使用癌细胞系中公开可获得的体外数据进行。使用独立的蛋白质组学(CCLE),药物反应(GDSC和CTD2)和转录组学(DEPMAP)验证了这些发现。采用了10倍的交叉验证策略来评估MOSA模型在多个OMICS层中的可重复性。
包容性和道德
所有作者都致力于维护自然投资组合期刊所主张的研究伦理原则和包容性。
报告摘要
有关研究设计的更多信息可在 自然投资组合报告摘要链接到本文。
数据可用性
所有数据均从癌症depmap中组装而成,生成的合成数据集已在以下URL下存放在FigShare中:DEPMAP数据集:https://doi.org/10.6084/m9.figshare.24420580。https://doi.org/10.6084/m9.figshare.24420598。MOSA增强数据集和潜在表示:https://doi.org/10.6084/m9.figshare.24562765。MOSA具有重要性:https://doi.org/10.6084/m9.figshare.24473005。MOFA多派重建和潜在表示:https://doi.org/10.6084/m9.figshare.24420631。Mixomics Muldi-omics潜在表示:https://doi.org/10.6084/m9.figshare.25764408。移动糖尿病多词的重建和潜在表示:https://doi.org/10.6084/m9.figshare.25764438。参考Trastulla,L.,Noorbakhsh,J.,Vazquez,F.,McFarland,J。&Iorio,F。癌细胞系的质量和临床相关性的计算估计。
摩尔。
系统。生物。 18,E11017(2022)。
Garnett,M。J.等。系统鉴定癌细胞中药物敏感性的基因组标记。自然 第483章,570 -575(2012)。
Barretina,J。等。癌细胞系百科全书可以实现抗癌药物敏感性的预测建模。自然 第483章,603 607(2012)。
Behan,F。M。等。使用CRISPRâCas9屏幕优先考虑癌症治疗靶标。自然 第568章,511 516(2019)。
Tsherniak,A。等。定义癌症依赖图。细胞 170,564â576.e16(2017)。
Pacini,C。等。癌细胞依赖性的全面临床信息图和目标优先顺序框架。癌细胞 42,301â316.e9(2024)。
Wekesa,J。S.&Kimwele,M。通过深度学习方法进行疾病诊断,预后和治疗的多摩学数据整合的回顾。正面。热内特. 14,1199087(2023)。
Cai,Z.,Poulos,R。C.,Liu,J。&Zhong,Q。癌症多摩学数据集成的机器学习。科学 25,103798(2022)。
Argelaguet,R。等。单细胞分辨率时小鼠胃肠道的多词分析。自然 第576章,487 A 491(2019)。
Argelaguet,R。等。MOFA+:多模态单细胞数据综合整合的统计框架。基因组生物学。 21,111(2020)。
Gonã§alves,E。等。949人细胞系的泛伴奏蛋白质组学图。癌细胞 40,835â849.e8(2022)。
Eraslan,G.,西蒙(L.纳特。交流。 10,390(2019)。
Freeman,B。A。等。mirth:通过秩转化和协调的代谢物归类。基因组生物学。 23,184(2022)。
Argelaguet,R。等。多态因子分析 - 多摩斯数据集的无监督集成的框架。摩尔。系统。生物。 14,E8124(2018)。
Boehm,J。S。等。癌症研究需要更好的地图。自然 第589章,514年516(2021)。
Poulos,R。C.,Cai,Z.,Robinson,P.J.,Reddel,R。R.&Zhong,Q。生物标志物发现药物组合学的机会。蛋白质组学 23,E2200031(2023)。
Minoura,K.,Abe,K.,Nam,H.,Nishikawa,H。&Shimamura,T。Experters的混合物深层生成模型,用于对单细胞多组学数据进行集成分析。细胞代表方法 1,100071(2021)。
Cohen Kalafut,N.,Huang,X。&Wang,D。多模式插补和嵌入的联合变异自动编码器。纳特。马赫。英特尔。 5,631 - 642(2023)。
他,Z.等人。与MIDAS的单细胞多模式数据的镶嵌整合和知识传递。纳特。生物技术。 42,1594年1605(2024)。
Ghazanfar,S.,Guibentif,C。&Marioni,J。C.使用未共享特征稳定镶嵌的镶嵌单细胞数据集成。纳特。生物技术。 42,284 A292(2024)。
Ashuach,T。等。Multivi:多模式数据集成的深层生成模型。纳特。方法 20,1222â1231(2023)。
Lundberg,S.M。&Lee,S.-I。解释模型预测的统一方法。在神经信息处理系统的进展(编辑Guyon,I。等人)卷。30(Curran Associates,Inc.,2017年)。
Van Der Meer,D。等。临床前癌模型的临床,遗传和功能数据集的细胞模型Passports-A枢纽。核酸研究。 47,D923âD929(2019)。
Dwane,L。等。项目评分数据库:研究癌细胞依赖性和优先考虑治疗靶标的资源。核酸研究。 49,D1365 d1372(2021)。
伊奥里奥,F.等人。癌症中药物基因组相互作用的景观。细胞 166,740 A754(2016)。
Garcia-Alonso,L。等。转录因子活性增强了癌症药物敏感性的标志物。癌症。 78,769 - 780(2018)。
李,H.等人。癌细胞系代谢的景观。纳特。医学。 25,850 -860(2019)。
Picco,G。等。通过药理学和CRISPR-CAS9筛选评估的基因融合到癌细胞适应性的功能连锁。纳特。交流。 10,2198(2019)。
Gonã§alves,E。等。通过整合药理和CRISPR筛选,药物机制的发现。生物Rxiv,https://doi.org/10.1101/2020.01.14.905729(2020)。
Meyers,R。M。等。拷贝数效应的计算校正提高了癌细胞中CRISPR-CAS9必要性筛查的特异性。纳特。热内特. 49,1779年1784年(2017年)。
Zampieri,G.,Vijayakumar,S.,Yaneske,E。&Angione,C。机器和深度学习符合基因组规模的代谢建模。公共科学图书馆计算。生物。 15,E1007084(2019)。
Allesã线,R。L。等。通过生成深度学习模型发现 2 型糖尿病的药物组学关联。纳特。生物技术。 41,399 408(2023)。
Lotfollahi,M.,Wolf,F。A.&Theis,F。J. Scgen预测单细胞扰动响应。纳特。方法 16,715â721(2019)。
Dempster,J。M.,Krill-Burger,J.,Warren,A。&McFarland,J。基因表达比基因组学具有更大的预测体外癌细胞脆弱性的功能。Biorxiv, https://doi.org/10.1101/2020.02.21.959627(2020)。
罗哈特(F.Mixomics:用于OMICS特征选择和多个数据集成的R软件包。公共科学图书馆计算。生物。 13,E1005752(2017)。
辛格,A.等人。暗黑破坏神:一种识别多摩学测定的关键分子驱动因素的综合方法。生物信息学 35,3055â3062(2019)。
Poulos,R。C.等。实现大规模蛋白质组学的策略进行可再现的研究。纳特。交流。 11,3793(2020)。
Nusinow,D。P.等。癌细胞系百科全书的定量蛋白质组学。细胞180 ,387 A 402.E16(2020)。文章
一个 中科院一个 考研一个 考研中心一个 谷歌学术一个 Gonã§alves,E。等。癌症基因组拷贝数变异的广泛转录后衰减。
细胞系统。5 ,386 398.E4(2017)。文章
一个 考研一个 考研中心一个 谷歌学术一个 Wainberg,M。等。共同模块的全基因组图图将功能分配给未表征的基因。
纳特。热内特. 53,638â649(2021)。
Seashore-Ludlow,B。等。在大型小分子灵敏度数据集中利用连通性。癌症发现。 5,1210年1223(2015)。
Rees,M。G。等。将化学敏感性与基础基因表达相关联揭示了作用机制。纳特。化学。生物。 12,109 - 116(2016)。
Mo,Q。等。综合癌症基因组数据中的模式发现和癌症基因识别。过程。国家科学院。科学。美国 110,4245 A4250(2013)。
DU,J.-H.,CAI,Z。&Roeder,K。通过SCVAEIT进行单模式多模式的镶嵌整合和插补的鲁棒概率建模。过程。国家科学院。科学。美国 119,E2214414119(2022)。
Meng,C.,Helm,D.,Frejno,M。&Kuster,B。Mocluster:识别多个OMICS数据集的关节模式。J.蛋白质组研究。 15,755 - 765(2016)。
Menden,M。P.等。基于基因组和化学特性对癌细胞敏感性的机器学习预测。公共图书馆一号 8,E61318(2013)。
Shorthouse,D.,Bradley,J.,Critchlow,S.E.,Bendtsen,C。&Hall,B。A. A.癌细胞系代谢景观的异质性。摩尔。系统。生物。 18,E11006(2022)。
奥伦,Y.等人。循环癌症持续细胞来自具有不同程序的谱系。自然 第596章,576 582(2021)。
Campit,S。E.等。一个合奏代谢组 - 异构体相互作用网络识别表观遗传药物的代谢物调节剂。生物Rxiv,https://doi.org/10.1101/2023.02.27.530260(2024)。
刘X.-R。等人。G9A抑制剂UNC0638抑制了上皮性间质转变介导的细胞迁移,并在三阴性乳腺癌中浸润。摩尔。医学。代表。 17 号,2239 2244(2018)。
Du,L.,Xie,F.,Han,H。&Zhang,L。通过减少EMT信号传导来抑制胃癌细胞的恶性表型靶向SALL4。抗癌研究。 43,4389年4401(2023)。
Park,S。J。等。BIX02189通过直接靶向TGF-α型受体来抑制TGF-α1诱导的肺癌细胞转移。癌症快报。 第381章,314 A322(2016)。
Ojima,T.,Kawami,M.,Yumoto,R。&Takano,M。A549细胞中甲氨蝶呤诱导的细胞死亡和上皮 - 间质转变的差异机制。毒性。资源。 37,293 300(2021)。
Meng,Q。等。谷胱甘肽过氧化物酶1的废除通过激活ROS介导的Akt/gsk3î²/蜗牛信号传导来驱动胰腺癌的EMT和化学耐药性。癌基因 37,5843 - 5857(2018)。
Pan,L.-N.,MA,Y.-F.,Li,Z.,Hu,J.-A。&Xu,Z.-H。KRAS G12V突变通过人类非小细胞肺癌中的TGF-α/EMT信号通路上调PD-L1的表达。细胞生物学。国际。 45,795â803(2021)。
张,Y.等人。全基因组CRISPR屏幕将PRC2和KMT2D-Compass鉴定为对转移有不同贡献的不同EMT轨迹的调节剂。纳特。细胞生物学。 24,554 -564(2022)。
郝X.等人。Mixgen:一种新的多模式数据增强。arXiv https://doi.org/10.48550/arxiv.2206.08358(2022)。
刘,Z.等人。学习特征空间中的多模式数据增强。arxiv, https://doi.org/10.48550/arxiv.2212.14453(2022)。
Pereira,R。C.,Santos,M。S.,Rodrigues,P。P.&Abreu,P。H.审查缺少数据插补的自动编码器:技术趋势,应用和结果。贾尔 69,1255年1285(2020)。
伊普森(N. B.),Mattei,P.-A。&Frellsen,J。Not-Miwae:深层生成建模,缺失并非随机数据。arxiv, https://doi.org/10.48550/arxiv.2006.12871(2020)。
Chen,J。,Xu,Y.,Wang,P。&Yang,Y。缺少随机数据的深度生成归因模型。在第 32 届 ACM 国际信息与知识管理会议论文集316â325(计算机协会,纽约,纽约,美国,2023年)。https://doi.org/10.1145/3583780.3614835。Marques-Silva,J。&Huang,X。解释性不是游戏。
arxiv,https://doi.org/10.48550/arxiv.2307.07514 (2023)。Ghandi,M。等。
癌细胞系百科全书的下一代表征。自然 第569章,503 508(2019)。
Pacini,C。等。癌症遗传依赖性的综合跨研究数据集。纳特。交流。 12,1661(2021)。
Dempster,J。M。等。两个大型PAN-CASTER CRISPR-CAS9基因依赖数据集之间的一致性。纳特。交流。 10,5817(2019)。
杨,W.等人。癌症药物敏感性基因组学 (GDSC):用于发现癌细胞治疗性生物标志物的资源。核酸研究。 41,D955 d961(2013)。
帕斯克,A.等人。Pytorch:命令式高性能深度学习库。副词。神经信息。过程。系统。32,8026 - 8037(2019)。Sundararajan,M.,Taly,A。&Yan,Q。深网的公理归因。
在第34届机器学习国际会议论文集(编辑。70 3319 3328(PMLR,2017年)。
Smilkov,D.,Thorat,N.,Kim,B.,Viã©Gas,F。&Wattenberg,M。Smoothgrad:通过添加噪声来消除噪音。arxiv, https://doi.org/10.48550/arxiv.1706.03825(2017)。
Asperti,A。和Trentin,M。差异自动编码器中平衡重建错误和kullback-leibler差异。IEEE接入 8,199440 199448(2020)。
Kingma,D。P.&Welling,M。自动编码的变分贝叶斯。arxiv, https://doi.org/10.48550/arxiv.1312.6114(2013)。
Kingma,D。P.&Welling,M。变分自动编码器的简介。arxiv, https://doi.org/10.48550/arxiv.1906.02691(2019)。
Akiba,T.,Sano,S.,Yanase,T.,Ohta,T。&Koyama,M。Optuna:下一代超参数优化框架。在第25届ACM SIGKDD国际知识发现与数据挖掘会议论文集2623 2631(计算机协会,2019年)。https://doi.org/10.1145/3292500.3330701。Mo,Q。等。
多型贝叶斯的全贝叶斯潜在变量模型,用于集成聚类分析多类型OMICS数据。生物统计学 19,71 - 86(2018)。
Ruepp,A。等。Corum:哺乳动物蛋白质复合物的综合资源。核酸研究。 36,D646 d650(2008)。
Chatr-Aryamontri, A. et al. The BioGRID interaction database: 2015 update.核酸研究。 43, D470–D478 (2015).
Szklarczyk,D.等人。The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible.核酸研究。 45, D362–D368 (2017).
伊奥里奥,F.等人。Unsupervised correction of gene-independent cell responses to CRISPR-Cas9 targeting.BMC基因公司 19, 604 (2018).
致谢
We thank the Broad Institute and the Wellcome Sanger Institute for, through the Cancer Dependency Map consortium, making their data freely available and readily accessible to the scientific community and thereby enabling this work. This research was funded in part by the Wellcome Trust Grant 206194. ProCan® is supported by the Australian Cancer Research Foundation, Cancer Institute New South Wales (NSW) (2017/TPG001,REG171150), NSW Ministry of Health (CMP-01), The University of Sydney, Cancer Council NSW (IG 18-01), Ian Potter Foundation, the Medical Research Futures Fund (MRFF-PD), National Health and Medical Research Council (NHMRC) of Australia European Union grant (GNT1170739, a companion grant to support the European Commission’s Horizon 2020 Program, H2020-SC1-DTH-2018-1,’iPC- individualized Paediatric Cure’ [ref. 826121]), and National Breast Cancer Foundation (IIRS-18-164). Work at ProCan® is done under the auspices of a Memorandum of Understanding between Children’s Medical Research Institute and the U.S. National Cancer Institute’s International Cancer Proteogenome Consortium (ICPC), that encourages cooperation among institutions and nations in proteogenomic cancer research in which datasets are made available to the public. Z.C. is the recipient of a PhD Scholarship from Sydney Cancer Partners with funding from Cancer Institute NSW (2021/CBG0002). A.R.B. is funded by the Portuguese national agency Fundação para a Ciência e a Tecnologia (FCT) through the research grant UI/BD/154599/2022. This work has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 951970 (OLISSIPO project). For open access, the authors have applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. This work was supported by national funds through FCT, under project UIDB/50021/2020 (https://doi.org/10.54499/UIDB/50021/2020). The authors acknowledge the OSCARS project, funded by the European Commission’s Horizon Europe Research and Innovation Programme under grant agreement No. 101129751.
道德声明
利益竞争
AstraZeneca, GlaxoSmithKline, and Astex Pharmaceuticals have awarded M.J.G.research grants and M.J.G.is founder and advisor at Mosaic Therapeutics.所有其他作者均声明不存在竞争利益。
同行评审
同行评审信息
自然通讯thanks Yejin Kim, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.同行评审文件可用。
附加信息
出版商的注释施普林格·自然对于已出版的地图和机构隶属关系中的管辖权主张保持中立。
补充资料
权利和权限
开放获取本文根据知识共享署名 4.0 国际许可证获得许可,该许可证允许以任何媒介或格式使用、共享、改编、分发和复制,只要您对原作者和来源给予适当的认可,并提供链接到知识共享许可证,并指出是否进行了更改。本文中的图像或其他第三方材料包含在文章的创意共享许可证中,除非在材料的信用额度中另有说明。如果文章的创意共享许可中未包含材料,并且您的预期用途不得由法定法规允许或超过允许的用途,则需要直接从版权所有者那里获得许可。要查看此许可证的副本,请访问http://creativecommons.org/licenses/by/4.0/。转载和许可
引用这篇文章

Cai, Z., Apolinário, S., Baião, A.R.
等人。Synthetic augmentation of cancer cell line multi-omic datasets using unsupervised deep learning.纳特·康姆15 , 10390 (2024). https://doi.org/10.1038/s41467-024-54771-4下载引文
:2023 年 12 月 21 日
:2024 年 11 月 18 日
:2024 年 11 月 29 日
:https://doi.org/10.1038/s41467-024-54771-4https://doi.org/10.1038/s41467-024-54771-4