
用于预测人血清蛋白与血液透析膜材料之间相互作用亲和力能量的机器学习模型
作者:Abdelrasoul, Amira
血液透析治疗是终末期肾脏疾病(ESRD)的常见治疗
1,,,,2。尽管血液透析疗法可以增强症状并延长患有ESRD的个体的寿命,但会导致并发症和不良反应3,,,,4,,,,5。这种负面后果可能包括疲劳,头痛,恶心,呕吐和低血压6,,,,7。此外,血液透析治疗的长期后果可能包括感染,心血管疾病,神经损伤,糖尿病,甚至死亡率8,,,,9,,,,10,,,,11。已经证明,在血液透析中使用更好的膜材料可以提高治疗的效率,并有助于减少与ESRD相关的并发症11,,,,12,,,,13,,,,14,,,,15。透析膜的血流相容性是关键因素,因为兼容性差会导致不良反应,例如血栓形成和溶血,损害患者的安全性和治疗结果13,,,,14,,,,16,,,,17,,,,18,,,,19。为了评估这些透析膜的血流相容性,研究人员经常测量亲和力能量20,,,,21,,,,22,它反映了膜和生物成分(例如蛋白质和细胞)之间相互作用的程度。但是,这种能量受膜表面的各种理化特性的影响,包括疏水/亲水平衡,静电力,表面粗糙度和电荷13,,,,23,,,,24。疏水表面倾向于通过与蛋白质的非极性区域相互作用来促进蛋白质吸附,从而增加亲和力25。相反,亲水性表面可以通过形成水合层来降低蛋白质吸附26。静电力也起着重要作用,因为膜的表面电荷和蛋白质上的电荷分布会导致吸引或排斥。例如,带负电荷的表面可能排除类似带电的蛋白质,但吸引了带积极的蛋白质。表面粗糙度和孔径大小通过提供蛋白质的其他结合位点进一步影响相互作用景观,从而增强蛋白质附着和亲和力能量27。膜表面上的官能团,例如羧基或胺基,可以与蛋白质形成特定的共价或非共价相互作用,从而影响吸附的强度和特异性26。因此,亲和能量是一种综合度量,反映了这些表面特性对膜血液相互作用的累积影响。虽然它是理解血流相容性的宝贵指标,但它涵盖了与膜的物理化学特性有关的多个因素28。此外,与传统的实验测试相比,计算方法已成为评估血流相容性的关键工具,从而可以进行更有效和具有成本效益的评估。除了了解这些表面相互作用外,测量设备与血液之间相互作用的强度对于确定使用过程中潜在的血流相容性问题至关重要。该评估可以帮助预测和减轻并发症,例如蛋白质结垢或血栓形成,以确保生物医学设备的安全有效性能20,,,,29,,,,30,,,,31。因此,开发具有较高血压相容的生物医学设备是一个复杂的过程,涉及一系列因素,包括材料选择,设计优化和在现实条件下进行测试11,,,,13,,,,32,,,,33,,,,34。尽管对医疗设备的仔细评估和测试可以帮助识别和解决开发过程中的潜在问题,但这可能是一个耗时,能源密集和昂贵的过程。因此,计算方法已成为有效设备评估的宝贵工具20,,,,35,,,,36,,,,37,,,,38,,,,39,,,,40,,,,41。分子对接是通常用于计算生物医学设备设计中亲和力能量的最常见计算方法。使用分子对接,研究人员可以根据材料的亲和力与血液成分的亲和力来预测材料的血流相容性26,,,,42。通过在设计过程的早期确定设备的亲和力,研究人员可以评估其血流相容性,并进行调整和修改以提高其安全性和功效43,,,,44。评估膜血流相容性分子对接也是一个耗时的过程,需要在对接前使用多种软件工具进行准备阶段。这些工具包括ChemDraw,Pymol,Biovia和Autodock Vina,需要化学专业知识,生物化学和生物信息学45,,,,46,,,,47,,,,48。
我们的研究团队已广泛研究了人血浆蛋白作为大分子受体和膜模型的作用。通过使用分子对接技术检查各种配体与人血清蛋白之间的结合亲和力相互作用,我们能够在进行实验研究之前准确地评估膜的血液相容性14,,,,15,,,,31,,,,49,,,,50,,,,51。
幸运的是,随着机器学习的最新进展52,,,,53,,,,54,,,,55,现在可以开发可以在相对较短的时间内准确预测亲和力的算法。开发这种算法的好处是多种多样的。首先,它将大大简化生物医学设备的设计过程56,,,,57,,,,58。研究人员可以快速测试并迭代其设计,从而导致更快,更有效的开发周期。此外,这些算法可以使研究人员能够设计具有更精确控制亲和力能量的设备,这可能会导致更有效和有针对性的治疗方法。
因此,我们的研究旨在开发一种强大的机器学习算法,以根据分子对接产生的全面数据集预测亲和力。为了实现这一目标,我们首先收集了一个分子对接结果的数据集,其中包括有关膜材料各种配体的结合亲和力能量的信息,这些膜材料具有不同特征,化学和对靶蛋白的方向的结合能量。该模型的新颖性源于其对不同化学结构的全面性和鲁棒性。我们测试了几种机器学习算法,包括线性回归,KNN回归,支持向量机,决策树回归,随机森林回归,XGBoost回归和Lasso回归,以评估其在亲和力能量预测中的性能。我们还调整了每种算法的超参数以优化其性能。总体而言,这项研究代表了应用机器学习方法的重要一步,以准确预测血液透析膜和其他生物医学应用所需的材料的血流相容性。结合亲和力能量对于识别潜在的候选药物至关重要。
材料和方法
数据收集和数据集说明
这项开创性的研究介绍了精心制作的数据集,这是我们研究小组勤奋工作的产物。该数据集涵盖了各种聚合物材料,包括原始聚合物,超氧化和亲水性化合物,环状聚合物以及阴离子和阳离子和阳离子。它还以各种比率和方向为ZW修饰的聚合物,以及伪ZW修饰的聚合物,接头,聚多巴胺和肝素。凭借其916个条目(表S.1。补充材料),我们的数据集不仅提供了一个全面的存储库来研究亲和力的复杂动力学,而且每个条目也有条不紊地设计为可行的实验验证。这种特征增强了其作为创新生物医学设备的知情设计的基础资源的价值,在该设计中,亲和力是一个关键的考虑因素。
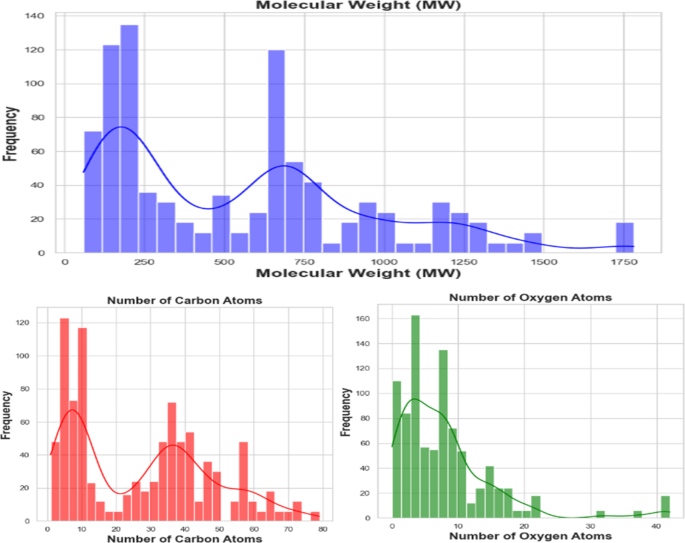
该数据集以其显着的多样性为特色,跨越了一系列化学化合物和聚合物,反映了亲和力研究的多维性质。该数据集包含广泛的成分,包括不同化学成分的原始聚合物,包括PES,PES,PSF,PVDF,PAN,PAN,PAN,纤维素单乙酸,纤维素二酰二乙酸和纤维素三乙酸酯等。它进一步扩展到结合各种接头,以其不同的长度和官能团为特色。在这一数据中,有一系列的zwitterions,跨越磷酸蛋白酶,硫贝因和羧贝因等,每种都会有助于分子相互作用的复杂网络。此外,这个全面的数据集涉及离子多样性领域,封装了许多阳离子和阴离子,每个阳离子和阴离子都具有其独特的特征。它大胆地研究了超疏水和亲水性涂层之间的对比,从而阐明了表面特性对亲和能量的深刻影响。包括肝素,聚多巴胺,环状和线性PMEA,以及与PES,PSF,PVDF和纤维素等改良的聚合物一起,与不同的Zwitter和表面涂层整合在一起,进一步增强了数据集的丰富性和复杂性。该数据集的一个关键特征是仔细准备各种聚合物与阳离子和阴离子的组合,从而有助于其深度,并对发挥作用的分子相互作用提供了全面的看法。此外,数据集还包括对PES和PSF的两个块和三块裂解的修改的亲和力值,提供了一个独特的机会来分析不同组合比的影响。图 1说明了关键分子描述子的分布,包括分子量,碳原子的数量和氧原子数。这些特征突出了配体的化学多样性,在血液透析膜技术的背景下提供了相关聚合物材料及其修饰的代表性观点。

分子量的分布,碳原子的数量,氧原子的数量。
受体蛋白选择的人血清白蛋白(HSA),纤维蛋白原β链(FB),转铁蛋白(TRF),血红蛋白(HEM),P2Y12嘌呤能受体和热休克蛋白(HSP)是血液透析(HD)的关键(HD)和膜研究,由于它们参与了血膜相互作用,这会影响血液相容性。HSA是最丰富的血浆蛋白,其吸附到透析膜上可能导致结垢,从而使其成为膜研究的关键重点。FB是血液凝结的核心,其与膜的相互作用可能引起血栓形成,这是HD的常见并发症。最大程度地减少FB吸附对于改善膜血流相容性至关重要。参与铁运输的TRF可以影响透析过程中的氧化应激,因此了解其与膜的相互作用有助于开发更多的生物相容性材料。研究在溶血期间释放的血红蛋白进行了研究,以减少透析膜引起的细胞损伤。涉及血小板聚集的P2Y12受体,可帮助研究人员设计抗直接膜,对于防止凝块形成至关重要。研究了与细胞应激反应有关的HSP,研究了其在炎症和生物相容性中的作用。通过严格的分子对接技术得出的精确亲和力计算的结合可显着提高数据集的范围和可靠性。该数据集标志着该领域的一个显着里程碑,代表了针对血液透析膜的第一个系统收集,这是如此广泛而多样化的亲和力数据。这个全面的数据集在亲和力能量预测领域内建立了一种新颖且动态有效的标准,提供了一种精简且有效的方法,可以在仅几秒钟内准确预测亲和力,这可以在新的见解和数据可用时进行更新。尽管配体是根据与血液透析膜技术相关性选择的,但我们确保数据集还包括新颖的化合物和化学修饰。这种方法有助于减轻对既定材料的任何潜在偏见,并提供分子相互作用的全面视图。为了进一步确保多样性,我们包括各种组合中的际离子修饰,阴离子/阳离子基团和疏水/亲水性化合物,提供了广泛的化学空间进行分析。
机器学习预测建模
配体蛋白数据集的分子建模
在构建我们的数据集的广泛过程中,该数据集由916行组成,我们做出了巨大的努力,以涵盖对HD膜修饰至关重要的各种材料。我们的数据集精心包括原始聚合物,各种ZWS,接头,伪ZW,肝素,超氧化物,亲水性化合物,环状聚合物,阴离子,阳离子,如图2所示。 2和附录1。这种全面的分类证明了我们的实质性奉献精神,从而确保了数据集在适应各种修改可能性方面的弹性。这种详尽的方法的重要性在为更准确的预测模型建立强大的基础上,从而扩大了我们的研究成果的精确性和可靠性。

HD膜修改的综合数据集概述。
这个关键阶段始于配体在数据集中包含的配体的彻底准备,涉及几个基本的程序步骤。这个过程涵盖了几个关键的过程步骤15,,,,42,,,,49:
-
1。
配体结构生成:使用化学绘制软件精确地执行配体结构的产生。随后,使用Chem3D Ultra进行了能量最小化,从而优化了配体结构以实现稳定且能量良好的构象。
-
2。
文件格式转换:这些优化的分子结构最初以摩尔格式的转换为PDBQT格式是随后的命令步骤。这种转换是使用多功能自动售货工具无缝执行的,以确保与下游分子对接软件的兼容性。
-
3。
分子对接:使用Autodock Vina Software 4.0版,可以促进分子对接实验的传导,这是创建数据集的关键的分子对接实验。该高级软件启用了每个配体 - 蛋白质相互作用的亲和力能量的计算。
作为分子对接研究的关键先决条件,通过从RCSB蛋白数据库(PDB)中检索3D X射线结构来认真制备蛋白质结构。这些蛋白质结构对于我们的研究是必不可少的,包括我们研究中心的关键蛋白,包括带有PDB代码1AO6的HSA,带有PDB代码3GHG的FB,带有PDB代码1D3K的TRF,以及带有PDB Code的HEM,PDB Code 2HHB,P2Y12,P2Y12,P2Y12,以及带有PDB代码5MKR的HSP。图 3说明了从各种配体 - 蛋白质相互作用的分子对接模拟获得的亲和力值的正态分布。使用标准化参数进行分子对接,每个受体的网格盒配置为完全覆盖结合位点。详尽水平为15来确保对结合姿势进行彻底探索。选择每种相互作用的最低结合能姿势以代表最稳定的配体 - 受体复合物,从而确保整个数据集的一致性。亲和力能量值遵循正态分布,范围从10到-2 kcal/mol,峰围绕7 kcal/mol,表明最常见的结合亲和力。较低的值代表更强的结合相互作用。直方图覆盖了核密度估计值(KDE),显示了平滑而对称的曲线,反映了亲和力能量的正态分布。

亲和力能量值的分布。
机器学习(ML)算法
通过分子对接技术完成数据集后,使用机器学习(ML)算法来预测亲和力。应用了多种ML算法,包括线性回归,k-nearest邻居(KNN)回归,决策树回归,随机森林回归,XGBoost回归,套索回归和支持向量回归。选择回归模型,以捕获自变量之间的复杂关系的能力(包括:蛋白质,原子数,碳原子数量,氮原子数量,磷原子的数量,氧原子数,氧原子的数量,硫原子的数量,分子量,分子量(MW),氢键供体的数量,氢键受体的数量,芳香环的数量,带电组的数量)和目标变量(亲和力能量),从而促进了对亲和力的准确预测,即连续数值参数。该数据集分为培训和测试集,总共包含916个记录。对于我们的预测建模,我们使用RDKIT库和来自6种不同蛋白质的数据从数据点提取了12个不同的参数,从而增强了模型的全面性和预测能力(Table' 1)。统计分析
为了评估开发模型的性能,我们采用了良好的指标,包括确定系数R平方(Râ²),均方误差(MSE)和平均绝对误差(MAE)。
这些指标是比较各种ML算法在我们数据集中预测亲和力能量方面的有效性的强大基础。
平均绝对误差(MAE)提供了预测值和实际值之间平均绝对偏差的度量。较低的MAE值表示更好的模型准确性,而较高的MAE值表明与地面真相值相比,模型的预测具有更大的平均误差。
$$ {\ text {mae}} = \ left({1/{\ text {n}}}}} \ right)*{\ text {s}}} \ left |{{{\ text {y}} - {\ hat {\ text {y}}}}}}} \ right | $ $
其中MAE是平均绝对误差,n是数据集中数据点的总数,| y -Å·|每个数据点的实际观察值(y)与预测值(Å·)之间的绝对差。
平方平方误差(MSE)提供了预测值和实际值之间平均平方偏差的度量。由于平方,它更加强调更大的错误,使其对异常值敏感。MSE值越低,模型越准确。MSE的公式如下:
$$ {\ text {mse}} =(1/{\ text {n}})*\ sum {({\ text {y}}} - \ hat {\ text {y} {y}})2}} $$
其中MSE是平均误差,n是数据集中数据点的总数(y -Å·)^2,是实际观察值(y)和预测值(Å·)之间的平方差。每个数据点。
R平方测量因变量中的方差比例,该变量由回归模型中的自变量解释。它提供了有关模型对观察到的数据拟合程度的洞察力,其R平方值较高,表明自变量的拟合度和更大的解释能力。
结果与讨论
基线数据评估
这项研究的基础锚定在严格的策划数据集上,并精心组装,以代表与血液透析膜相关的广泛聚合物材料。由于我们的研究团队仔细而故意的综合,该数据集没有缺少值,包括916个独特的条目。每个条目都体现了独特的聚合物组成,修饰或涂料类型,可确保对强大分析亲和力分析所需的可变空间的全面覆盖。变量包括跨度定量分子描述符,例如分子量,各种原子类型的计数以及结构特征,以及描述聚合物类型和修饰的定性属性。在分析之前,将所有连续变量进行标准化,以确保可比性并减轻不同尺度的影响,尤其是针对广泛的分子量。此步骤对于避免任何不当偏见至关重要,以使机器学习算法可能施加更大的幅度特征。尽管通过代表各种化学家族和结构的代表来主动最小化数据集构建中的潜在偏见,但我们承认对特定聚合物的内在偏见,并根据其在血液透析应用中的普遍性和重要性所选择的修改。该数据集的设计旨在反映血液透析膜技术的当前景观,并预测该领域的未来方向。然而,数据集的构建原理和归一化过程为随后的机器学习努力建立了坚实的基础,以确保数据准备应用高级分析技术和有意义的见解的推导。
导航亲和力能量预测
血液透析膜和各种生物医学设备在现代医疗保健中起关键作用,尤其是在治疗终末期肾脏疾病(ESRD)和其他威胁生命的状况方面。确保透析膜血流相容性至关重要,因为它们与血液的相互作用可能对患者的安全和福祉产生深远的影响。此外,无论是可植入的医疗设备,药物输送系统还是体外设备的其他生物医学应用的材料混血性,它们的成功都取决于它们与患者的循环系统无缝集成的能力。计算亲和力能量是评估这些设备的血流相容性的基本步骤。血液透析膜和其他生物医学设备必须与循环系统复杂的环境表现出和谐的共存,以确保患者的福祉,从而使亲和力的准确计算在其设计和评估中必不可少的先决条件。对分子对接的偏爱可以归因于其在没有任何限制的情况下预测每个配体和受体的亲和力能量的能力,以及配体之间相互作用的强度和性质,通常是生物医学装置表面和靶蛋白的组成部分在血液中发现。这种预测能力对于确定设备与循环系统的兼容性至关重要,因为不利的相互作用可能导致患者严重并发症。
分子对接过程通常是从使用ChemDraw等软件的配体结构产生的,然后进行能量最小化以实现稳定且能量良好的构象。随后,配体结构将转换为与分子对接软件(例如PDBQT)兼容的格式,这对于随后的对接过程至关重要。分子对接本身是使用Autodock Vina等专业软件执行的,该软件采用算法来预测最佳的结合姿势和亲和能量值。尽管分子对接是评估亲和力能源的强大工具,但它广泛依赖包括ChemDraw,Pymol,Biovia Discovery Studio和Autodock Vina在内的各种软件工具,使其成为了耗时且资源密集的过程。例如,配体和蛋白质结构的制备需要对细节的细致关注,从而在研究管道中消耗了宝贵的时间。此外,使用多种软件工具可以引入复杂性和潜在的错误来源,尤其是在处理大型数据集或复杂的分子结构时。在某些关键情况下,例如紧急医疗方案或时间敏感研究,全面分子对接分析所需的延长持续时间可能会带来重大的挑战。例如,如果患者需要快速治疗,涉及具有定制的血液相容性的血液透析膜,那么准确的亲和力计算所需的大量计算时间可能会及时干预。因此,尽管分子对接仍然是亲和力预测的黄金标准,但它可能并不总是与某些医疗保健或研究需求的紧迫性保持一致。还必须注意的是,在生物医学设备的背景下,对高分子量配体和受体的分子对接可能非常耗时。随着配体和受体的大小和复杂性,计算需求呈指数增长,有时需要几个小时甚至几天才能完成单个对接模拟。这种延长的处理时间可以进一步限制在迅速决策或实时结果必须进行的情况下使用分子对接的实用性。
在评估生物医学设备的血液相容性的情况下,几个基于用户友好的基于Web的平台,例如Swissdock和Ligdock等可访问的替代方案,提供优势和缺点。这些在线工具提供了一种简化的亲和力预测方法,与分子对接相比,通常需要更少的计算专业知识。但是,必须深入研究这些平台的细微差别,以了解它们的全部潜力和局限性。基于用户友好的网络平台(如Swissdock和Ligdock)的优势主要在于其可访问性和速度。这些平台使用户可以轻松地上传其分子结构,通常以蛋白质数据库(PDB)或PDBQT格式上传,并在几分钟内计算亲和力。这个简化的过程大大减少了启动亲和力计算所需的时间和精力。此外,这些平台是为用户友好性而设计的,使得它们可用于更广泛的研究人员和医疗保健专业人员,甚至是那些没有广泛计算背景的专业人员。但是,这些基于用户友好的Web平台有明显的缺点。一个明显的限制是对PDB或PDBQT格式的分子结构的要求,这意味着用户必须使用软件工具进行经常耗时的预处理步骤,以确保配体和受体结构的兼容性。尽管这消除了一些复杂性,但并不能完全消除对专业软件工具的需求。此外,这些平台通常对与亲和力计算有关的配体和受体的大小和复杂性施加限制。当试图评估大型,生物学相关的分子(例如血液蛋白(如白蛋白或纤维蛋白原))之间的相互作用时,这可能是尤其有限的,这些分子在血液相容性评估中起着关键作用。因此,无法检查这些大分子与生物医学装置表面之间的关键相互作用可能是一个重要的劣势。总而言之,基于用户友好的网络平台(如Swissdock和Ligdock)为计算亲和力能量提供了方便且耗时的手段,使其成为研究人员和医疗保健从业者的有吸引力的选择。但是,至关重要的是要认识到,它们并不完全消除对预处理的需求,并对所涉及的分子结构的大小和复杂性施加限制。这些局限性可能会影响亲和力评估的全面性,特别是在与大血蛋白相互作用至关重要的情况下。因此,尽管这些平台提供了有价值的可访问性,但研究人员应仔细权衡其优势和缺点,以确定其适合特定血流相容性评估任务的性。
鉴于这些考虑因素,用于预测亲和力能源的机器学习(ML)算法的开发和应用为传统分子对接技术提供了有希望的和创新的替代方案,以及Swissdock和Ligdock等基于网络的平台。ML算法的主要优点在于它们能够简化亲和力计算的能力,同时规避了与结构文件格式转换相关的费力预处理步骤(例如,PDB或PDBQT文件),这是分子对接的先决条件。此外,基于ML的模型消除了配体和受体的尺寸限制,这是某些基于Web的工具(例如Swissdock和Ligdock)的重要缺点。一旦训练,机器学习模型就可以快速,准确地预测亲和力能量,从而在时间限制至关重要的情况下极有效。与分子对接不同,这可能需要大量的计算资源和小时,以预测高分子重量配体和受体的亲和力能量,ML算法只需单击几下即可提供近乎实用的结果。当评估生物医学设备的血液相容性时,这种迅速的预测过程尤其有利,在这种情况下,快速决策对于患者的安全至关重要。此外,ML算法在研究环境中提供多功能性和适应性。这些模型可以通过新的数据集进行连续更新和改进,并结合了最新的实验发现并提高了它们的预测准确性。这种适应性可确保模型在不断发展的研究环境中保持相关性和有效性,从而有助于持续的血液透析膜和其他生物医学设备的完善。将ML算法整合到此类设备的研究和开发过程中,这是朝着更有效的和更有效的,和高效的和高效,和高效的,,,和开发数据驱动的医疗解决方案。
数据分析和机器学习模型选择
鉴于这些进步,我们的探索始于对为我们的机器学习应用程序提供信息的基础数据的深入分析。在深入研究模型性能的细节之前,必须了解初始数据特征及其对随后的分析步骤的影响。在图 4,相关图揭示了多种分子特征与亲和力能量之间的复杂关系,这是一个指示分子结合倾向的关键参数。这种全面的分析突出了一系列变量,这些变量显示出与绝对亲和力的相关性,其中大小和统计意义分别通过条形的高度说明并分别用星号标记。观察到芳族环数量的显着正相关,表明芳环计数的增加通常与较高的亲和力能量相关。这种关系可能受芳香族化合物固有的分子量和大小增加的影响,这会导致复杂的结构,这些结构可能引入空间障碍,改变电子分布并影响表面特性,从而增加结合亲和力。相反,protein_trâ,protein_p2y12和protein_fb-与亲和能量具有统计学上显着的负相关性。这些负相关可能表明与靶标的相互作用降低或降低结合亲和力,从而突出了这些蛋白质在结合动力学中的复杂性质及其对分子亲和力的潜在影响。此外,图形表示表明,氮原子数量和绝对亲和力能量之间存在边际相关性,这意味着氮原子计数对整体结合亲和力的影响可忽略不计。p值注释证实了相关性的鲁棒性,星号表示显着性水平,分别代表低于0.05、0.01和0.001的p值。这些发现共同强调了分子相互作用的复杂性质以及结构细微差别在决定分子行为和结合特性中的重要性。
框图(图 5a)所描绘的框图描述了分子内芳环结构的发生率与其亲和力能量之间的关联。在检查的数据集中,具有芳香环的实体倾向于表现出升高的分子量,更大的氧原子计数以及复杂的构象属性,这些属性与增强的中位数亲和力相吻合。这些分子的复杂结构组成可能会增强亲水性和疏水性相互作用,从而对结合亲和力产生相当大的影响。
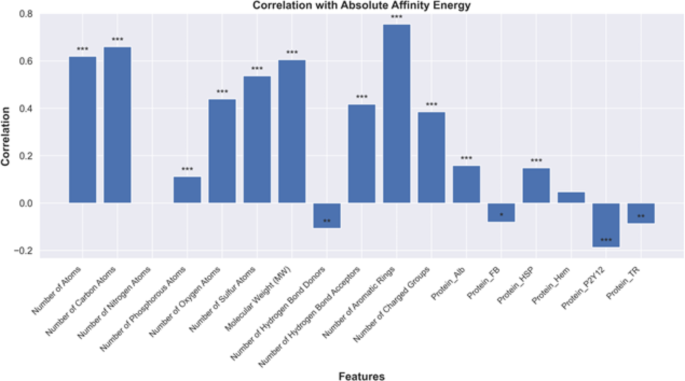
分子特征与亲和力之间的相关性:评估结构和蛋白质特征对结合倾向的影响。
图。 5B对三种特定蛋白的存在或不存在亲和力的比较观点,与早期有关其相互作用的发现相关。与缺乏相比,Protein_P2Y12和Protein_tr与中位亲和能量相比,中位亲和能量较低,与已建立的负相关与亲和力能量保持一致。值得注意的是,Protein_P2Y12与Protein_TR相比,在存在条件下与中位数表现出更大的偏差,从而增强其更强的负相关性,并表明对降低结合亲和力的影响更明显。相反,蛋白质下摆与亲和能的边际正相关,这是由于存在时中值的略有升高所证明的。这表明虽然蛋白质下摆的存在可能导致结合亲和力的增加,但效果相对微妙。重叠的四分位间范围和晶须的程度表明,尽管趋势是可观察到的,但数据存在很大差异,这可能是由于复杂的生物学相互作用或其他分子因子的影响而在此分析中未分离的。

((一个)Boxplot比较有或没有芳香环的分子的亲和力能量。((b)箱形图显示了与蛋白质_p2y12,hem和tr的存在有关的亲和力分布。为了确定预测亲和力的最熟练的机器学习模型,我们测试了各种回归算法,包括线性回归,KNN,决策树回归,随机森林回归,XGBoost,XGBoost,Lasso回归和支持矢量回归。
选择这些模型以涵盖广泛的复杂性,从而使我们能够评估每种方法如何处理数据集的特定特征。随机森林和XGBoost以其集合学习能力而闻名,特别适合这项任务,因为它们可以在数据中捕获复杂的非线性关系。这些模型由于能够减少过度拟合并在不同数据子集之间提供稳健性能的能力而优先考虑。这两种模型汇总了来自多个决策树的预测(在随机森林的情况下)或迭代地构建模型以最大程度地减少错误(对于XGBoost),从而使其对于具有复杂模式的数据集非常有效。相反,包括线性回归和拉索回归等较简单的模型,为性能和解释性提供了基准。这些模型使我们能够评估复杂性和准确性之间的权衡。尽管它们更易于解释和更快地训练,但它们可能缺乏捕获分子数据中存在的非线性相互作用的能力,这通常会限制其预测能力与集合方法相比。
根据关键指标(例如R)进行严格评估2,MAE和MSE,两个模型浮出水面,作为优越的竞争者:随机森林回归和XGBoost回归(图。 6)。随机森林和Xgboost回归的表现都非常出色,前者略微磨损了后者。r2得分是我们的预测符合实际值的程度。与r2得分为0.8987,随机森林模型解释了我们数据集中差异的约89.87%。该指标越接近1,模型的拟合度越好。相比之下,XGBoost回归得分为0.8368,表明它解释了大约83.68%的方差。我们还使用MSE和MAE评估了我们的模型。

基于Râ²,MAE和MSE性能指标的回归模型的比较评估。
尽管MSE给更大的错误提供了更大的重量,但可能突出了我们的模型表现特别差的实例,但MAE不论其方向如何,都可以更清楚地了解平均误差幅度。我们使用网格搜索和随机搜索策略对随机森林和XGBoost模型进行了广泛的高参数调整。对于随机森林,我们专注于优化参数,例如n_estimators(树的数量),max_depth(最大树深度),min_samples_split(拆分节点所需的最小样本数)和min_samples_leaf)。对于XGBoost,我们调整了包括Learning_rate(步骤尺寸收缩以控制过度拟合),N_estimators,MAX_DEPTH和子样本(用于训练每棵树的数据的一部分)的参数。在某些情况下,高参数调整导致了边缘改善,但它在防止过度拟合和确保强大的模型性能方面发挥了重要作用。诸如随机森林中的max_depth和min_samples_leaf之类的参数,以及XGBoost中的Learning_rate和子样本,有助于调节模型复杂性,从而最大程度地减少过度拟合并确保模型在训练和测试数据集中的表现良好。即使两个模型的结果都接近,XGBoost在随机森林后面表现出轻微的性能滞后。这种差异可能是由于数据集的固有结构所致,在该结构中,随机森林的合奏方法具有更简单的结构,非常适合捕获基础数据模式。我们的随机森林模型的MSE为0.36,MAE为0.45,表明与XGBoost的MSE相比,持续的性能始终取得更好的性能,MAE的MAE为0.4908。鉴于稍高的性能指标以及随机森林回归因其整体性质而抗拒过度拟合的先天能力,我们选出了随机森林回归作为亲和能量预测的主要模型。随机森林回归是一种合奏学习方法,在训练过程中创造了决策树的森林。每棵树都经过数据和功能的随机子集训练,使该模型适合噪声和过度拟合。通过平均多个树木的预测,随机森林自然会减少方差并改善概括。为了进一步解决对过度拟合的担忧,我们对所有模型采用了5倍的交叉验证,从而确保了数据的不同子集的一致性。与其他模型(例如XGBoost)相比,随机森林具有整体方法,表现出更好的概括,这些模型表现出更大的性能和对数据分割的敏感性的差异。跨数据集的随机森林的稳定结果突出了其坚固性和对过度拟合的抵抗力,使其成为此任务的更可靠模型。尽管XGBoost提供了强大的梯度增强框架,并且可以有效地处理各种数据类型和分布,但其复杂性和众多的超参数有时可以使其成为微不足道的模型,可以进行微调和解释。相比之下,随机森林具有更简单的超参数集和可解释的结构,可以更加直观的理解和更容易的优化,与我们的目标更好地保持一致,即找到一个简单地预测亲和力能量的模型。应该注意的随机森林模型的关键优势之一是其可伸缩性。随着越来越多的数据可用,尤其是在诸如分子对接之类的快速发展的领域中,该模型可以在没有大量大修的情况下有效地进行更新,从而确保其随着时间的推移保持相关和有效。此外,随机森林具有基于其重要性对特征进行对的能力,提供了宝贵的见解,该见解对分子特性对结合亲和力能量具有最大的影响。图 7。提出了从随机森林回归模型中得出的特征重要性,揭示了预测结合亲和力能量最重要的分子和原子特征。该分析不仅为模型性能的优化提供了信息,而且还为分子对接研究中的实验方法提供了宝贵的见解,从而使对蛋白质配体相互作用的潜在机制进行了更具针对性的研究。该图表明分子量(MW)是最重要的特征,突出了其对结合亲和力的重要影响。这表明,具有较高分子量的较大分子更可能建立更强的结合相互作用,这可能是由于可用于相互作用的表面积增加所致。同样,碳原子的数量和原子数量是高度影响力的特征,强调了分子大小和原子组成在确定结合强度中的作用。这些特征共同指出了分子大小,原子组成和结合亲和力能量之间的牢固关系。在重要特征的列表中,芳香环的数量在结合相互作用中起着关键作用。已知芳香环参与PI-PI堆叠相互作用,这对于稳定蛋白质配体复合物至关重要。氧原子的数量也显示出非常重要的,这反映了氧在形成氢键中的作用,这是分子相互作用中的关键因素。特定的蛋白质,例如蛋白质_p2y12,protein_hem和protein_hsp,也被强调为有影响力的特征。这些蛋白质的参与表明它们的存在或不存在显着影响结合亲和力,这反映了所建模的系统的生物学相关性。这些发现表明,分子对接预测不仅取决于原子特性,而且还受到特定蛋白质 - 配体相互作用的高度影响,这可能是实验数据集中的关键重点。尽管诸如氢键供体数量,氢键受体的数量和带电组的数量等特征的重要性较低,但它们在模型中仍然起着不可忽略的作用。这些特征反映了分子的静电特性,这是配体和蛋白质结合位点之间相互作用的基础。静电相互作用虽然在整体特征的重要性中并不占主导地位,但为了解分子环境如何影响亲和力的方式提供了必要的细节。最后,与特定原子元素有关的特征,例如磷原子的数量,硫原子的数量以及氮原子的数量,有助于模型预测,但与更广泛的结构和组成特征(如分子量和原子计数)相比,重要性较小。这表明虽然单个原子类型会影响结合相互作用,但在预测结合亲和力能量时,它们的作用是总体分子结构和组成的次要的。总而言之,此特征重要性分析不仅可以清楚地了解哪些分子特征最强烈影响结合亲和力能量,而且还提供了对这些特征在生物学和化学环境中如何相互作用的见解。这些发现强调了分子相互作用的多面性质,其中大小,组成和特定原子或蛋白质相关相互作用共同确定结合亲和力。

从随机森林模型中得出的特征重要性的层次分布用于预测结合亲和力能量。
在我们的机器学习模型(尤其是随机的森林回归模型)中追求预测精度时,采用了详尽的系统方法来优化模型的超参数,这是增强模型性能的过程关键。Recognizing that the default parameters often provide a suboptimal balance between the bias and variance, we engaged in advanced tuning techniques to meticulously refine these settings, aiming to bolster the model’s predictive accuracy and generalizability.Initially, a comprehensive Grid Search was conducted, an algorithm that iteratively explores a predefined array of hyperparameters to pinpoint the combination yielding the highest performance, based on the R-squared (R2) 分数。考虑到与每个附加参数的组合呈指数增长,这种方法在计算苛刻和时间密集型上是详尽的。通过采用5倍的交叉验证策略,进一步加强了这种方法的鲁棒性,其中训练集被分配为五个子集,并且该模型经过迭代培训和验证。This procedure not only ensured a comprehensive exploration of the hyperparameter space but also validated the model’s stability and performance across different data segments.To ensure the reproducibility of our results amidst the inherent stochastic nature of the Random Forest algorithm, we instantiated our model with a fixed random state (i.e., ‘random_state = 101’).This deliberate move aimed to control the random number generation used in the training process, thereby providing consistency in the model’s behavior and the subsequent hyperparameter exploration.为了规避网格搜索的计算局限性,我们随后采用了一种随机搜索策略,该方法在选择超参数时引入了一定程度的随机性。Unlike the Grid Search which exhaustively traverses through the entire parameter grid, the Randomized Search randomly samples a specified number of parameters sets from broader distributions, providing a balance between computational efficiency and the exploration of the optimal hyperparameter space.
Utilizing the ‘RandomizedSearchCV’ function from the Scikit-learn library, we specified extensive distributions for several critical hyperparameters.These included ‘n_estimators’, representing the number of trees in the forest, with a random integer distribution between 100 and 500;‘max_depth’, indicating the maximum depth of the trees, with values ranging from 10 to 50 (alongside the default ‘None’ option);‘min_samples_split’, specifying the minimum number of samples required to split an internal node;and ‘min_samples_leaf’, the minimum number of samples required at each leaf node.这些参数不是任意选择的,而是基于经验证据和理论考虑,以确保全面搜索。与网格搜索的确定性性质相比,这种方法的随机性使参数空间更加多样化和广泛检查。我们启动了100个迭代的随机搜索,每次都采用5倍的交叉验证策略来维持验证鲁棒性,从而增强了发掘更有效和细微的超参数配置的可能性。Despite these rigorous optimization techniques, the enhancements in the model’s predictive performance metrics post-tuning were relatively marginal, highlighting the model’s sensitivity to hyperparameter adjustments.Specifically, the improvements in MSE and the R2score were not substantial, underscoring the complex interplay between the model’s structure and the dataset’s characteristics.性能增强的平稳性强调了机器学习模型优化的复杂,有时难以捉摸的性质,并非所有调整都会产生重大改进。It also highlights the potential influence of inherent data characteristics and feature dynamics on the model’s learning efficacy.These insights suggest that while hyperparameter tuning is indisputably essential, it is but one component in a constellation of factors, including data quality, feature engineering, and inherent algorithmic characteristics, that collectively dictate a model’s ultimate predictive capability.
Validation of machine learning algorithm
It’s crucial to emphasize that the ligands under consideration during the validation were not part of the modeling development.The model’s predictions extended beyond its training set, showcasing its versatility in predicting affinity energy for molecular entities that were not explicitly part of the initial model development.Moreover, it is important to note that the selected proteins, such as Protein_TR, were chosen for illustrative purposes, and the model’s capacity extends to predicting affinity energy for any polymer interacting with any protein.这种灵活性强调了该模型作为在生物医学设备设计和评估背景下预测各种分子相互作用的结合能的可靠工具的广泛适用性和潜力。The ligands listed in Table 2, including PSF-CB-PB (2:1 combination ratio of ZW), PSF-SB-PB (2:1 combination ratio of ZW), Coating layer of Vinyl Sulphonic Acid (VSA) + 2-(Dimethylamine)ethyl methacrylate (PDMAEMA) as a Pseudo ZWs layer, PDMAEMA Cation, Acrylic acid + PDMAEMA as a Pseudo ZW Coating layer, and PES-Pseudo ZW as a grafting combination, represent new combinations that were not included in the dataset used for model training. All these ligands are feasible to be experimentally verified, further demonstrating the innovative approach and practical applicability of our model in exploring novel biomaterial interactions.
The novelty of the model stems from its comprehensiveness and robustness for different chemical structures, as it predict any new chemical structure outside of the dataset. Expanding the utility of our robust machine learning framework, we proceeded to apply the optimized Random Forest model to an uncharted set of molecular entities to predict their binding affinity energies. This phase was instrumental in evaluating the model’s real-world applicability and precision in forecasting affinity energy outside the confines of the training data. The new data, comprising a diverse array of molecular structures, presented a quintessential test bed for our computational approach. The predictions generated by the Random Forest model were meticulously compared with results obtained from traditional molecular docking studies. This comparative analysis was crucial in not only validating the machine learning model’s predictive prowess but also in highlighting its efficiency and potential advantages over more conventional techniques. The comparison of the two methodologies is summarized in Table 2, which delineates the predicted affinity energies by the Random Forest model against the corresponding values derived from molecular docking procedures. Interestingly, our results showed substantial agreement between the two sets of results, supporting the model’s capability of predicting binding energy with a high degree of accuracy and consistency. This alignment highlights the effectiveness of the Random Forest model in mimicking the complex processes involved in molecular interactions, which are usually studied using molecular docking techniques. FB plays a central role in hemodialysis membrane design, influencing membrane performance and hemocompatibility crucial for blood purification. Fibrinogen is a key protein involved in blood coagulation and fibrin formation. Its importance lies in its role in maintaining hemostasis, preventing excessive bleeding, and promoting wound healing. In the specific context of hemodialysis membranes, FB plays a critical role in molecular interactions, influencing the performance and hemocompatibility of the membrane. Its presence and interaction significantly impact factors such as thrombosis and fouling, which are essential considerations for the efficacy of hemodialysis in blood purification. Understanding and addressing the influence of FB are crucial for designing membranes that integrate seamlessly with the circulatory system, ensuring optimal performance and minimizing the risk of complications during medical procedures. Our group has correlated FB adsorption with increased inflammation and elevated levels of clotting factors. Additionally, it plays a critical role as it competes with other proteins to adsorb onto the membrane surface from the mixture, as demonstrated in our recent studies using synchrotron in-situ imaging59,,,,60。For this protein, computing affinity energy in traditional molecular docking takes a lot of time due to its large size, but our machine learning model predicts affinity energy for FB complexes in seconds.This expedited capability, essential in critical healthcare scenarios, circumvents the time-consuming nature of conventional methods.However, it is worth noting a specific observation concerning the Protein_TR feature.The Random Forest model assigns importance values to each feature based on how frequently a feature is used to split data in the trees and how well those splits improve the model’s accuracy.From the Feature Importances plot (Fig. 7), it is evident that the Protein_TR feature does not rank very high in terms of importance. This suggests that, during the training phase, the Random Forest did not utilize this feature extensively to make decisions. Moreover, the inclusion of this feature did not substantially enhance the model’s predictive accuracy. This could potentially account for the variations observed when using the model to predict affinity energy for Protein_TR as compared to other proteins. While the difference in predicted affinity energy for Protein_TR between this model and molecular docking is not exceedingly large, it is more pronounced than for other proteins. Notably, the Random Forest model exhibited a remarkable predictive accuracy, with deviations from the molecular docking results falling within an acceptable range, thereby asserting its reliability. This attests to the model’s robustness and its potential utility as a reliable tool in predicting binding energies.
The comparison, as detailed in Table 2, reveals a remarkable concordance between the computational predictions of the Random Forest model and the molecular docking results, with error percentages ranging narrowly from 0 to 10%. This minor deviation highlights the Random Forest model’s robust predictive capability, substantiating its utility as a viable alternative to the molecular docking simulations. Notably, the low percentage error affirms the model’s precision in anticipating the affinity energy for novel polymer combinations, spanning various orientations and ratios, with and without the integration of linkers. It effectively captures interactions across a diverse protein panel, encompassing Human Serum Albumin (HSA), Fibrinogen (FB), Transferrin (TR), Hemoglobin (Hem), Heat Shock Protein (HSP), and P2Y12.
结论
In this study, we ventured into the realm of computational biology, specifically focusing on the innovative application of machine learning algorithms to predict molecular affinity energies, a domain traditionally governed by molecular docking methods. Affinity energy, a critical parameter in the interface of biomolecular interactions, holds paramount significance in the design and application of biomedical devices, particularly in assessing fouling resistance. Therefore, controlling affinity energy is the key to reducing biofouling, enhancing device performance, and ensuring patient safety. In certain exigent scenarios, such as the rapid optimization of hemodialysis membranes to enhance biocompatibility and performance, the development of targeted therapeutic interventions or the swift screening of potential drug candidates during a health crisis, the ability to swiftly assess affinity energies becomes crucial. Traditional molecular docking approaches, while thorough, are notoriously time-consuming and computationally intensive, particularly when analyzing large ligand-receptor complexes, posing challenges in situations where time is of the essence. Conversely, machine learning models stand out in these critical junctures, offering rapid, precise predictions of affinity energies with just a few clicks. Given the pivotal role of affinity energy in biomedical applications, we elected to harness the capabilities of ML for its prediction, in particular to streamline the optimization process in hemodialysis membrane design. Our approach utilized a comprehensive dataset comprising 916 entries, encompassing a diverse spectrum of components such as pristine membrane polymers, linkers, cations, anions, hydrophobic and hydrophilic compounds, heparin, Zwitterions, and various specially modified membranes which are prevalent in hemodialysis membrane modification. Traditionally, the affinity energies of these compounds have been ascertained through molecular docking, a method known for its accuracy but also its computational exigency, particularly in the analysis of extensive, varied datasets. By applying machine learning algorithms to this rich repository of data, we sought to transcend the limitations of molecular docking, enabling rapid, efficient predictions of affinity energy. These models bypass the exhaustive computations by leveraging historical data and learned patterns, thereby providing immediate insights that are especially vital in guiding early-stage drug discovery and expediting the materialization of medical solutions. Proceeding with the analytical rigor of this study, we embarked on a comparative evaluation of multiple regression methodologies to ascertain the most efficacious predictive model for molecular affinity energies. This comparative analysis encompassed a spectrum of machine learning models, including linear regression, KNN regression, decision tree regression, random forest regression, XGBoost regression, lasso regression, and support vector regression. Each model was meticulously trained and tested using a robust dataset of 916 records, with the prediction models harnessing 12 distinct parameters extracted from the data points alongside 6 unique proteins. The efficacy of the developed models was critically assessed through key statistical metrics: R2, MSE, and MAE. Our findings delineated that the random forest model exhibited superior performance with an R2 of 0.8987, MSE of 0.36, and MAE of 0.45, indicating a high level of predictive accuracy and reliability on the training dataset. Concurrently, the XGBoost model demonstrated commendable predictive capabilities with an R2of 0.83, MSE of 0.49, and MAE of 0.49. Notably, while both models presented robust performances, the random forest model surpassed its counterparts in accuracy on the testing dataset, underscoring its predictive precision and the robustness of its algorithmic structure.The diagnostic plots showcased provide a testament to the efficacy of our Random Forest model. A foundational strength of the Random Forest algorithm is its capacity to manage complex data structures, address potential over-fitting through bagging and averaging and discern nonlinear patterns. This prowess is evident in the Residual Histogram (Fig. 8
A), where residuals are predominantly centered around zero, indicating that the model’s predictions are, on average, unbiased. The bell-shaped distribution further underscores that the model’s errors are random and not systematic, a desirable characteristic suggesting that the model captures most of the patterns in the data without being overly influenced by noise, and the normality assumption is largely held. Furthermore, the Q-Q Plot of Residuals provides additional clarity on the distribution of residuals (Fig. 8b)。The central region of the plot shows a close alignment of the data points with the red theoretical line, which reinforces the notion of normality and illustrates the model’s ability to handle the bulk of the data efficiently.The slight deviations in the tails, while drawing attention to potential outliers, also emphasize the model’s sensitivity in detecting extreme values without being overly swayed by them.

Diagnostic Plots for Random Forest Regression Model: (一个) Residual Histogram and (b) Q-Q Plot.
The congruence between the predicted outcomes and established biochemical benchmarks not only validates the Random Forest model as a reliable predictive tool but also illuminates its potential as a transformative asset in streamlining drug discovery pipelines and facilitating more nuanced investigations into bio-molecular affinities. Moreover, the model afforded a rapid, efficient, and cost-effective alternative to the often time-consuming and resource-intensive molecular docking simulations, without compromising on accuracy. This efficiency is particularly advantageous in the preliminary screening of vast molecular libraries, expediting the identification of promising compounds for subsequent in-depth analysis.
The success of the Random Forest model in this application reinforces its position as a formidable tool in the field of drug discovery and bioinformatics research. By demonstrating comparable, if not superior, performance to established molecular docking methods, the model stands out as a valuable asset for future studies focused on the exploration of molecular binding affinities, offering a blend of precision, efficiency, and broad applicability.
As we draw from the findings of our research, it becomes evident that the journey of enhancing the predictive power of machine learning models, particularly in molecular affinity predictions, has just begun. While the Random Forest model has demonstrated commendable capabilities in predicting affinity energies, it is important to recognize the model’s current constraints and the need for its continuous evolution in line with the dynamic nature of computational biology. The model’s accuracy is intrinsically tied to the dataset’s quality and scope used for its training, which, although diverse, reflects only a snapshot of proposed polymer-protein interactions. Our model has yet to be tested against the vast diversity and scale of molecular datasets found in real-world scenarios. Thus, enhancing the model’s adaptability and scalability remains imperative. Collaborative efforts that integrate pharmacological, computational biological, and data science insights are essential for refining the model’s robustness and maintaining its applicability in various contexts. For future work, we recommend a multifaceted approach to model improvement: Firstly, expanding the dataset to encompass a broader spectrum of interactions, including those with rare or newly discovered compounds, would likely increase the model’s generalizability. Additionally, real-world validation is necessary—though the model’s predictions align with molecular docking results, experimental verification remains the definitive measure of utility. We should rigorously test the model’s predictions experimentally, thereby establishing its efficacy beyond computational environments. Moreover, assessing the model against a wider array of proteins and polymers can broaden its practical utility. To this end, regularly incorporating new data and discoveries will be critical for the model’s continued relevance, especially in the rapidly progressing field of biomaterials. In conclusion, our work highlights the significant potential of integrating computational techniques with biological insights. This intersection not only facilitates advancements in biomedical device design but also promises to forge new frontiers in bioinformatics and related disciplines.
数据可用性
The data required to reproduce these findings are available from the corresponding author (A. Abdelrasoul) upon reasonable request.
参考
Busink, E. et al. A systematic review of the cost-effectiveness of renal replacement therapies, and consequences for decision-making in the end-stage renal disease treatment pathway.欧元。J. Health Econ。, 1–16 (2022).
El-Zaatari, Z. M. & Truong, L. D. Renal cell carcinoma in end-stage renal disease: A review and update.生物医学 10, 657 (2022).
文章一个 PubMed一个 PubMed Central一个 数学一个 Google Scholar一个
Alostaz, M., Correa, S., Lundy, G. S. & Waikar, S. S. & Mc Causland, F. R. Time of hemodialysis and risk of intradialytic hypotension and intradialytic hypertension in maintenance hemodialysis.J. Hum。高血压。, 1–11 (2023).
Canaud, B. et al. Hidden risks associated with conventional short intermittent hemodialysis: A call for action to mitigate cardiovascular risk and morbidity.World J. Nephrol. 11, 39 (2022).
文章一个 PubMed一个 PubMed Central一个 数学一个 Google Scholar一个
Derk, G. et al. The safety and efficacy of clonidine in hemodialysis patients: A systematic review and Meta-analysis.药理, 1–11 (2022).
Hamal, S. S. & Khadka, P.Acute Complication Dur. Hemodial.(2023)。
Stern, J. I. et al. Narrative review of migraine management in patients with renal or hepatic disease.Headache: J. Head Face Pain。63, 9–24 (2023).文章
一个 数学一个 Google Scholar一个 Chang, J. F. et al. A joint evaluation of impaired cardiac sympathetic responses and malnutrition-inflammation cachexia for mortality risks in hemodialysis patients.正面。
医学7 , 99 (2020).文章
一个 广告一个 数学一个 Google Scholar一个 Genovesi, S. et al. Atrial fibrillation and morbidity and mortality in a cohort of long-term hemodialysis patients.是。
J.肾脏51 , 255–262 (2008).文章
一个 PubMed一个 数学一个 Google Scholar一个 Usui, N. et al. Association of cardiac autonomic neuropathy assessed by heart rate response during exercise with intradialytic hypotension and mortality in hemodialysis patients.肾脏int。
101, 1054–1062 (2022). 文章一个
PubMed一个 数学一个 Google Scholar一个 Nazari, S. & Abdelrasoul, A. Surface zwitterionization of hemodialysismembranesfor hemocompatibility enhancement and protein-mediated anti-adhesion: a critical review.Biomedical Eng.
ADV。, 100026 (2022).Mollahosseini, A., Abdelrasoul, A. & Shoker, A. A critical review of recent advances in hemodialysis membranes hemocompatibility and guidelines for future development.
母校。化学物理。 248, 122911 (2020).
文章一个 Google Scholar一个
Nazari, S. & Abdelrasoul, A. Impact of membrane modification and surface immobilization techniques on the hemocompatibility of hemodialysis membranes: A critical review.膜 12, 1063 (2022).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Saadati, S. et al. In situ synchrotron imaging of human serum proteins interactions, molecular docking and inflammatory biomarkers of hemocompatible synthesized zwitterionic polymer coated-polyvinylidene fluoride (PVDF) dialysis membranes.冲浪。接口。27, 101505 (2021).文章
一个 数学一个 Google Scholar一个 Westphalen, H. et al. Case studies of clinical hemodialysis membranes: influences of membrane morphology and biocompatibility on uremic blood-membrane interactions and inflammatory biomarkers.科学。
代表。10 , 14808 (2020).文章
一个 广告一个 PubMed一个 PubMed Central一个 Google Scholar一个 Abdelrasoul, A. & Shoker, A. Induced hemocompatibility of polyethersulfone (PES) hemodialysis membrane using polyvinylpyrrolidone: Investigation on human serum fibrinogen adsorption and inflammatory biomarkers released.化学
工程。res。des。 177, 615–624 (2022).
文章一个 Google Scholar一个
Eduok, U., Abdelrasoul, A., Shoker, A. & Doan, H. Recent developments, current challenges and future perspectives on cellulosic hemodialysis membranes for highly efficient clearance of uremic toxins.母校。Today Commun. 27, 102183 (2021).
文章一个 Google Scholar一个
Radu, E. R., Voicu, S. I. & Thakur, V. K. Polymeric membranes for biomedical applications.聚合物 15, 619 (2023).
文章一个 PubMed一个 PubMed Central一个 数学一个 Google Scholar一个
Wei, Q., Feng, S. & Wu, L. Glomerular endothelium-inspired anticoagulant surface coating on polyethersulfone hemodialysis membrane.母校。化学物理。,,,,127364(2023)。
Mollahosseini, A. & Abdelrasoul, A. Novel insights in hemodialysis: most recent theories on the membrane hemocompatibility improvement.Biomedical Eng.ADV。, 100034 (2022).
Mollahosseini, A., Saadati, S. & Abdelrasoul, A. Effects of mussel-inspired coâ€deposition of 2â€hydroxymethyl methacrylate and poly (2â€methoxyethyl acrylate) on the hydrophilicity and binding tendency of common hemodialysis membranes: molecular dynamics simulations and molecular docking studies.J. Comput。化学 43, 57–73 (2022).
文章一个 PubMed一个 Google Scholar一个
Song, X., Ji, H., Zhao, W., Sun, S. & Zhao, C. Hemocompatibility enhancement of polyethersulfone membranes: Strategies and challenges.ADV。膜。 1, 100013 (2021).
文章一个 数学一个 Google Scholar一个
Dubovskii, P. V. & Efremov, R. G. The role of hydrophobic/hydrophilic balance in the activity of structurally flexible vs. rigid cytolytic polypeptides and analogs developed on their basis.Expert Rev. Proteomics。15, 873–886 (2018).文章
一个 PubMed一个 Google Scholar一个 Arumugam, S., Chwastek, G. & Schwille, P. Protein–membrane interactions: The virtue of minimal systems in systems biology.Wiley Interdiscip.
Rev.: Syst.生物。医学 3, 269–280 (2011).
数学一个 Google Scholar一个
Nazari, S. & Abdelrasoul, A. Surface zwitterionization of hemodialysismembranesfor hemocompatibility enhancement and protein-mediated anti-adhesion: A critical review.生物。工程。ADV。 3, 100026 (2022).
文章一个 Google Scholar一个
Nazari, S. & Abdelrasoul, A. Simulation-based assessment of zwitterionic pendant group variations on the hemocompatibility of polyethersulfone membranes.功能。Compos.母校。 5, 12 (2024).
文章一个 PubMed一个 PubMed Central一个 数学一个 Google Scholar一个
Woo, S. H., Park, J. & Min, B. R. Relationship between permeate flux and surface roughness of membranes with similar water contact angle values.9月纯净。技术。 146, 187–191 (2015).
文章一个 数学一个 Google Scholar一个
Lalia, B. S., Kochkodan, V., Hashaikeh, R. & Hilal, N. A review on membrane fabrication: structure, properties and performance relationship.淡化 326, 77–95 (2013).
文章一个 Google Scholar一个
Bonhoure, A. et al. Benchtop holdup assay for quantitative affinity-based analysis of sequence determinants of protein-motif interactions.肛门。生物化学。 603, 113772 (2020).
文章一个 PubMed一个 数学一个 Google Scholar一个
Mollahosseini, A. & Abdelrasoul, A. Molecular dynamics simulation for membrane separation and porous materials: A current state of art review.J. Mol。图形。模型。 107, 107947 (2021).
文章一个 PubMed一个 数学一个 Google Scholar一个
Mollahosseini, A., Saadati, S. & Abdelrasoul, A. A. Comparative assessment of human serum proteins interactions with hemodialysis clinical membranes using molecular dynamics simulation.大分子。ther。模拟。 31, 2200016 (2022).
文章一个 Google Scholar一个
Jain, S. & Parashar, V. Critical review on the impact of EDM process on biomedical materials.母校。制造。过程。36, 1701–1724 (2021).文章
一个 数学一个 Google Scholar一个 Wadhera, T., Kakkar, D., Wadhwa, G. & Raj, B. Recent advances and progress in development of the field effect transistor biosensor: A review.J. Electron.
母校。48 , 7635–7646 (2019).文章
一个 广告一个 数学一个 Google Scholar一个 Li, Y., Liu, X., Liu, H. & Zhu, L. Interfacial adsorption behavior and interaction mechanism in saponin–protein composite systems: A review.Food Hydrocoll.
136, 108295 (2023). 文章一个
数学一个 Google Scholar一个 Chen,Z。等。Alternatingly amphiphilic Antimicrobial oligoguanidines: Structure–property relationship and usage as the Coating Material with unprecedented hemocompatibility.
化学母校。 34, 3670–3682 (2022).
文章一个 数学一个 Google Scholar一个
Dacrory, S., Hashem, A. H. & Hasanin, M. Synthesis of cellulose based amino acid functionalized nano-biocomplex: Characterization, antifungal activity, molecular docking and hemocompatibility.环境。Nanatechnol.监视。管理。 15, 100453 (2021).
Das, S. et al. Microwave-assisted β-cyclodextrin/chrysin inclusion complexation: An economical and green strategy for enhanced hemocompatibility and chemosensitivity in vitro.J. Mol。Liq。 310, 113257 (2020).
文章一个 数学一个 Google Scholar一个
Liu, Y., Xie, N., Tang, Y. & Zhang, Y. Investigation of hemocompatibility and vortical structures for a centrifugal blood pump based on large-eddy simulation.物理。流体。34, 115111 (2022).文章
一个 广告一个 Google Scholar一个 Romanova, A. N. & Telyshev, D. V. inIV International Conference on Control in Technical Systems (CTS).
135–137 (IEEE).(2021)。Zambrano, B. A. et al. Computational investigation of outflow graft variation impact on hemocompatibility profile in LVADs.
艺术品。器官。48, 375–385 (2024).文章
一个 PubMed一个 数学一个 Google Scholar一个 Fischer, L. et al. Impact of extracorporeal blood pump gap sizes on the performance and hemocompatibility under off-design operation.艺术品。
器官(2024)。Forli, S. et al. Computational protein–ligand docking and virtual drug screening with the AutoDock suite.
纳特。原始 11, 905–919 (2016).
文章一个 PubMed一个 PubMed Central一个 数学一个 Google Scholar一个
Tiwari, G., Kumar, S., Chauhan, M. S. & Sharma, D. Ab-initio, molecular docking and md simulation of an anti-hiv drug (lamivudine): An in-silico approach.生物。母校。开发。2, 1002–1016 (2024).文章
一个 数学一个 Google Scholar一个 Shen,D。等。Molecular docking-guided design on glucose-responsive nanoparticles for microneedle fabrication and three-meal-per-day blood-glucose regulation.
ACS应用。母校。接口。15, 31330–31343 (2023).文章
一个 PubMed一个 Google Scholar一个 Jadhav, S. A., Sen, D. B., Sen, A. K. & Shah, A. P. In silico pharmacokinetics and docking analysis of active biomolecules from 5-amino-salicylic acid against cyclin dependent kinase II.NeuroQuantology
20, 364 (2022).Beg, M. & Athar, F. Pharmacokinetic and molecular docking studies of Achyranthes aspera phytocompounds to exploring potential anti-tuberculosis activity.J. Bacteriol。
Mycol.打开。使用权。 8, 18–27 (2020).
数学一个 Google Scholar一个
Ravelliani, A. et al. Study of Molecular Docking on compounds with potential as anti-inflammatory.Jurnal Eduhealth。13, 1070–1078 (2022).
数学一个 Google Scholar一个
Benjamin, I. et al. Antimalarial potential of naphthalene-sulfonic acid derivatives: Molecular electronic properties, vibrational assignments, and in-silico molecular docking studies.J. Mol。结构。 1264, 133298 (2022).
文章一个 Google Scholar一个
Saadati, S. et al. Assessment of polyethersulfone and polyacrylonitrile hemodialysis clinical membranes: In situ synchrotron-based imaging of human serum proteins adsorption, interaction analyses, molecular docking and clinical inflammatory biomarkers investigations.母校。Today Commun. 29, 102928 (2021).
文章一个 数学一个 Google Scholar一个
Saadati, S. et al. Biocompatibility enhancement of hemodialysis membranes using a novel zwitterionic copolymer: Experimental, in situ synchrotron imaging, molecular docking, and clinical inflammatory biomarkers investigations.母校。科学。Engineering: C。117, 111301 (2020).文章
一个 数学一个 Google Scholar一个 Sishi, Z. et al. Influence of clinical hemodialysis membrane morphology and chemistry on protein adsorption and inflammatory biomarkers released: In-situ synchrotron imaging, clinical and computational studies.生物。
工程。ADV。 5, 100070 (2023).
文章一个 数学一个 Google Scholar一个
Al-Kharusi, G., Dunne, N. J., Little, S. & Levingstone, T. J. The role of machine learning and design of experiments in the advancement of biomaterial and tissue engineering research.生物工程 9, 561 (2022).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Hafeez, M. A. et al.在。8th International Conference on Information Technology Trends (ITT).154–159 (IEEE).(2022)。
Krishnan, R., Rajpurkar, P. & Topol, E. J. Self-supervised learning in medicine and healthcare.纳特。Biomedical Eng., 1–7 (2022).
Monaghan, C., Looper, K. & Usvyat, L. inTechnological Advances in Care of Patients with Kidney Diseases17–23Springer, (2022).
Tettey, F., Parupelli, S. K. & Desai, S. A review of biomedical devices: Classification, regulatory guidelines, human factors, software as a medical device, and cybersecurity.生物。母校。设备。2, 316–341 (2024).文章
一个 Google Scholar一个 Goh, G. D. et al. Machine learning for bioelectronics on wearable and implantable devices: Challenges and potential.组织工程。
A部分。29, 20–46 (2023).文章一个
PubMed一个 数学一个 Google Scholar一个 McDonald, S. M. et al. Applied machine learning as a driver for polymeric biomaterials design.纳特。
社区。14 , 4838 (2023).文章
一个 广告一个 PubMed一个 PubMed Central一个 数学一个 Google Scholar一个 Abdelrasoul, A., Westphalen, H., Kalugin, D., Doan, H. & Shoker, A. In situ synchrotron quantitative analysis of competitive adsorption tendency of human serum protein to different clinical hemodialysis membranes and assessment of potential impacts.Biomedical Eng.
ADV。6 , 100104 (2023).文章
一个 Google Scholar一个 Abdelrasoul, A., Zhu, N., Doan, H. & Shoker, A. In-situ synchrotron quantitative analysis of competitive adsorption tendency of human serum proteins on polyether sulfone clinical hemodialysis membrane.科学。
代表。13 , 1692 (2023).文章
一个 广告一个 PubMed一个 PubMed Central一个 Google Scholar一个 下载参考致谢
作者信息
作者和隶属关系
道德声明
竞争利益
作者没有宣称没有竞争利益。
道德批准
The principal investigator of the project, Dr. Amira Abdelrasoul, has the Research Ethics Approval and the Operational Approval to conduct the research in the Saskatchewan Health Authority, in Canada. She has responsibility for the regulatory approvals that pertained to this project. All experimental protocols involving humans were conducted according to the governing law.
附加信息
Publisher’s note
关于已发表的地图和机构隶属关系中的管辖权主张,Springer自然仍然是中立的。
权利和权限
开放访问This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material.您没有根据本许可证的许可来共享本文或部分内容的改编材料。The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.要查看此许可证的副本,请访问http://creativecommons.org/licenses/by-nc-nd/4.0/。重印和权限
引用本文

Nazari, S., Abdelrasoul, A. Machine learning models for predicting interaction affinity energy between human serum proteins and hemodialysis membrane materials.
Sci代表15 , 3474 (2025). https://doi.org/10.1038/s41598-024-83674-z下载引用
:2024年9月24日
:2024年12月16日
:2025年1月28日
:https://doi.org/10.1038/s41598-024-83674-z关键字