
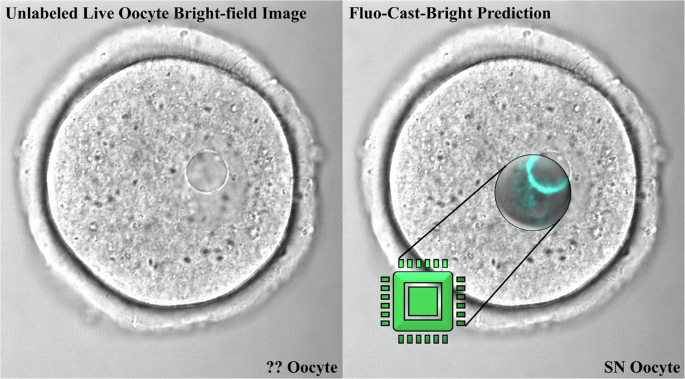
Fluo-cast-Bright:一种深度学习的管道,用于无创染色质结构和活卵母细胞中发育潜力的预测
作者:De La Fuente, Rabindranath
介绍
哺乳动物细胞的核被组织成功能专业的隔室,这些室是控制基因表达和分化所需的。在相间细胞核中,将异染色质结构域组织为高度凝结,转录抑制性核聚集体或镀铬对准确的染色体分离和维持基因组稳定性至关重要1,,,,2。卵母细胞核,也称为生发囊泡(GV),表现出独特的染色质构型,在产后卵母细胞生长的最后阶段受到动态修饰。哺乳动物前卵母细胞中染色质结构的功能分化的特征是核体系结构的醒目变化为减数分裂开始。这个过程是通过大规模染色质重塑的机制进行的,并且是表观遗传成熟的关键组成部分,以赋予具有减数分裂和发育潜力的完全生长的卵母细胞所需1,,,,2。在人和小鼠的卵母细胞中,最初存在反应的染色质构型,以支持合成和累积对减数分裂成熟至关重要的母体mRNA储存所需的高水平的全局转录。3,,,,4,,,,5。在这些卵母细胞中,异染色质域或染色体缺乏与核仁的任何关联,导致形成非弹性的核仁(NSN)构型6。原位杂交和与主要卫星DNA探针可视化异染色质结构域的现场杂交和活细胞成像表明,染色体与核仁逐渐相关,并随后大规模的染色质重塑导致形成核糖周围的核糖rim,导致到周围的核核酸核酸核核酸核酸核酸核酸核酸核酸核酸核酸核酸核酸核酸核酸核酸核酸核酸核酸核酸核酸纤维核的形成(SN)为减数分裂开始准备的配置6,,,,7,,,,8。这个关键的发展里程碑还伴随着卵母细胞基因组中新生转录活性的全球抑制3,,,,4,,,,5。
大规模染色质结构的遗传和药理学操纵都表明,将染色质重塑为SN构型可能会提供上十分位异染色质,并具有及时减数分裂进展所需的功能构型和准确的染色体隔离6。与这个概念一致,NSN卵母细胞表现出异常的减数分裂成熟,并且经常在体外受精后不超出2细胞阶段9,,,,10,,,,11。因此,积累的证据表明,将染色质重塑为SN构型对于赋予具有全减数分裂和发育潜力的哺乳动物卵母细胞至关重要1,,,,6,,,,9,,,,10,,,,11。
识别卵母细胞质量和发育潜力的细胞或分子标记是紧迫的需求,并且是在人类不育症和动物再生产领域实施临床和研究应用至关重要的一步。但是,到目前为止,对发育潜力的任何潜在可靠细胞标记物进行检测和/或定量需要侵入性程序,例如蛋白质交联和免疫荧光分析单个卵母细胞限制任何后来的下游应用。
现在,机器学习的最新进展使得可以开发计算深度神经网络,并具有从未标记的细胞中未经标记的微观图像识别关键信息的能力,而没有事先侵入性分析,这些细胞对人眼并不明显12,,,,13。计算神经网络,例如无标签成像13和类似的方式,例如硅标签12代表细胞功能的非侵入性分析的范式转移,因为它们可以实时从相比或传输光图像中获取有关细胞器和细胞子结构的定量信息12,,,,13。在这里,我们利用了最先进的方法在深度学习网络开发中的分析能力,以预测未标记的小鼠前卵巢卵母细胞中染色质构型和发育潜力的状态。我们针对高发育潜力的非侵入性选择的卵母细胞的策略集中在生发囊泡阶段高度分辨的染色质构型的荧光图像预测以及在赖氨酸9(H3K9ME3)上的组蛋白三甲基化的定位,这是我们的表观遗传标记(H3K9ME3),这是一种表观遗传标记。维持细胞分裂期间染色体结构14,两个真正与卵母细胞减数分裂和发育潜力直接相关的善意标记。
我们的方法将荧光预测网络与分类步骤相结合,以告知染色质状态和基因组完整性。核分割后,使用训练有素的深神经网络提取染色质信息,以预测来自明亮场图像的染色质荧光图。然后,通过三个分类网络扫描荧光图,以确定卵母细胞是否表现出NSN或SN构型。我们的管道在固定卵母细胞的分类中达到了91.3%的精度和活卵母细胞分类的85.7%精度。机器学习工具最近已开发用于分析哺乳动物配子的工具15,,,,16。但是,据我们所知,这是专为使用无标签,明亮的田间图像的实时哺乳动物卵母细胞中染色质结构和发育潜力的非侵入性预测而设计的第一条神经网络管道。我们的深度学习选择管道在几分钟之内提供有关卵母细胞染色质状态的重要信息快速有效。作为概念的证明,我们证明了其在具有高发育潜力的卵母细胞的非侵入性选择和下游转录分析中的应用,从而产生了独特的减数分裂和发育能力胜任的卵母细胞的数据集对染色质结构和/或卵母细胞转录组产生不利影响。
结果
开发深度学习管道,用于在排卵前卵母细胞中对染色质构型的非侵入性预测
哺乳动物卵母细胞的活细胞成像提供信息丰富的信息,相比或传输光显微照片,其中卵母细胞核以及核仁的大小,位置和结构信息很明显(图很明显) 1a)。但是,如果没有先前的侵入性免疫荧光分析核结构,则无法检测到每种卵母细胞上存在的染色质构型的类型(图 1B)。因此,我们开发了一条深度学习的管道,可以准确预测来自明亮场,活卵母细胞图像的染色质构型的荧光图像,然后生成NSN/SN分类。我们的管道可以有效地施放大型染色质结构的荧光预测,并在明亮场图像上高分辨率,从而可以无创选择活卵母细胞。重要的是,基于SN卵母细胞的准确荧光预测和鉴定,我们的管道不仅可以辨别染色质结构,而且还可以精确地选择具有高度减数分裂和发育潜力的活卵母细胞。

一个共聚焦显微镜下GV级卵母细胞的明亮场图像。封闭核(蓝色虚线)的感兴趣区域(平方)被分割并用于分析染色质构型。指示核仁的位置(星号)。b在从NSN到SN构型的过渡过程中,卵母细胞在核仁周围获得了独特的染色质边缘(星号),这与发育和减数分裂潜力的增加有关,并且与转录活性降低有关。侵入性荧光标签是目前确定染色质构型的标准实践,如使用H3K9ME3免疫化学(RED)和DAPI染色质标记(蓝色)所示。NSN卵母细胞表现出反应的染色质构型,而SN卵母细胞显示出紧凑的染色质结构和围绕核仁的明亮染色质边缘。
我们的结构fluo恢复预测和cl作为Sificat离子开布里gHT - 场(Fluo-Cast-Bright)管道17如图所示 2。我们的管道最初是在蛋白质交联后使用固定卵母细胞对核结构进行训练的。一堆明亮的场共聚焦图像用于生成整个卵母细胞核或生发囊泡的3-D重建,以进行精确的核分割(图。 2a)。为了训练我们的荧光预测网络,我们使用了两个目标标签DAPI和H3K9me3的相应的3-D像素注册图像(所有通道代表相同的空间定位)来确定存在的染色质构型类型在每个卵母细胞上(图 2a)。

一个荧光预测网络是基于UNET ++结构构建的,并使用3-D明亮场图像作为输入以及DAPI(DNA)和H3K9me3的组合荧光图作为目标标签进行训练。b分类网络是基于Resnext101体系结构构建的,并使用来自最佳荧光预测网络的预测荧光图作为输入,将正确的NSN/SN标签作为目标标签进行培训。cFluo-cast-Bright管道的工作流程使用具有未知染色质构型的卵母细胞的3-D明亮场图像堆栈。
为了设计荧光预测模型,我们使用了具有三维U-NET ++的卷积神经网络(CNN)18建筑(图 2a和补充图 1a)用于图像分割和翻译任务,能够从高级图像特征中提取信息,例如核形态以及低级特征,例如线条和边缘,并生成一个使用相对位置和图像的全面解释系统识别和细分对象并发现图像翻译的隐藏功能的功能18。跳过途径进一步增强了这种体系结构,这使得低级特征与对图像的高级了解以提高性能18(补充图 1a)。为了实现荧光预测,与地面真相或实际结合DAPI的荧光图相比和H3K9me3在生发囊泡中的定位(图 2b)。我们的分类网络基于Resnext-10119建筑(图 2b;补充图 1B),其中包含身份快捷连接,使复杂的计算分析能够改善图像分类19。然后,使用3-D预测的荧光图的2-D图像从荧光预测模型中产生的3-D图像来区分NSN和SN卵母细胞(图。 2b)。2-D预测的荧光图和每个图像的手动注释的NSN/SN分类用于训练分类网络(图。 2b)。
这两个体系结构的选择是根据在许多图像分析任务中的八个不同模型体系结构和特定参数进行直接比较,以及它们在预测染色质状态方面的最佳功效。与其他常用体系结构相比 2a)。此外,与我们的荧光图分类任务中的其他常用体系结构相比,Resnext-101结构具有更高的分类精度(补充图。 2b)。重要的是,高参数测试还证明了我们模型中使用的最佳参数集(补充图。 2C)。
训练我们的荧光预测和分类模型管道(图 2a和b),我们使用了138个固定卵母细胞的3-D明亮场和荧光图像堆栈,其中96个用于训练,其中42个用于验证。在用于验证的图像上具有最佳性能的模型被指定为最佳模型,然后使用三种不同的火车磁导板拆分来训练从最佳模型产生的42个验证图像的荧光图。我们使用3-D图像来训练荧光预测模型和2-D图像来训练分类模型(图 2C)。虽然不同的2D横截面之间的关系有助于荧光预测,但单个二维横截面包含足够的信息,以将卵母细胞分类为NSN或SN。因此,我们从每个3-D预测的荧光图堆栈(总共672,2-D荧光图)中使用了16,2-D荧光图,以训练三个分类模型(图。 2b和c)。根据其验证性能选择最佳分类模型。
为了测试我们非侵入性分类方法的功效,我们在23个固定卵母细胞的未标记的明亮场图像上测试了我们的Fluo-ast-Bright管道。将单个卵母细胞转移到玻璃底96孔板的单独标记的井中,并一次拍照,以仔细跟踪每个卵母细胞。使用我们的管道分析了这些明亮的卵母细胞图像,将NSN或SN预测的标签分配给每个卵母细胞(图 2C)。在初始计算分类之后,使用DAPI染色用于对相同固定卵母细胞的单个分析,以揭示使用共聚焦显微镜的每个卵母细胞的实际染色质构型NSN或SN。预测的染色质状态与实际染色质状态(或地面真相)之间的比较表明,正确预测了23个卵母细胞中21种的染色质构型(准确性= 91.3%;图5;图。 3a)。然后,我们对固定卵母细胞进行了第二次测试。这次,我们使用玻璃底培养皿对23个固定和未标记的卵母细胞进行了成像,并将固定的卵母细胞转移到矿物油下的Mem培养基(50-¼L)的微滴中,以模拟活卵母细胞成像的条件。通过DAPI染色和共聚焦显微镜确定的计算预测的染色质状态与实际染色质状态之间的比较表明,正确预测了23个卵母细胞中21分的染色质构型(准确性= 91.3%)。重要的是,对三个其他预测指标的分析显示出类似的高值(Precision = 91.4%,回忆= 96.4%,F1得分= 93.8%),表明表现的性能是我们的神经网络可在固定卵母细胞中的不同成像条件下转移,并仅使用明亮的场图像准确预测染色质状态。

条图显示了固定卵母细胞上浮动涂层管道的分类性能(n= 2个独立测试)。b在成像,分割和非侵入性分类之前,实时GV阶段卵母细胞池。c条形图显示了活卵母细胞上浮动式亮点管道的分类性能(n= 2个独立测试)。d使用Fluo-Cast-Bright管道将明亮场图像的分段核区域和染色质构型分类为NSN或SN。e分类的卵母细胞可用于下游应用或固定并标记以确定管道的准确性。相应的dapi(青色)标签的明亮场图像(如(所示)d)用于染色质状态的地面真相验证。除非划分(红色X)的所有卵母细胞都可以通过Fluo-Cast-Bright管道准确地预测。
在活前卵母细胞中的染色质构型的无创预测
接下来,我们使用活卵母细胞测试了我们的氟铸造管道的预测准确性(图。 3b),因为这对于我们管道的广泛研究和临床应用至关重要。活细胞成像揭示了卵母细胞核的微妙(尽管可检测到的运动)存在。因此,由于在活卵母细胞中亚细胞成分的细胞内移动,我们的流量 - 饰面模型的预测准确性降低。尽管这些运动可能会影响荧光预测和分类步骤,但我们发现,仅用活卵母细胞对分类网络进行重新训练足以提高管道的准确性,因为网络能够识别运动的效果在预测的荧光图中,并在分类步骤中消除了它。我们使用来自63个活卵母细胞产生的明亮场图像堆栈完成了此重新训练三个分类模型。根据验证性能的结果再次选择最佳的分类模型。
重新训练分类网络后,我们使用42个新的活卵母细胞图像进行了测试,该图像未参与任何培训或重新训练过程。通过管道分析了未标记的明亮场图像,以基于实时卵母细胞图像的非侵入性计算分析来生成初始的NSN或SN分类。在产生预测后,DAPI染色和共聚焦显微镜揭示了实际的染色质状态或地面真相。该测试是在每次重复21个活卵母细胞的两个独立实验重复中进行的(图 3b)在2天的时间内收集。预测的染色质状态与实际染色质地面真理之间的比较表明,正确预测了21个卵母细胞中18种的染色质构型(准确性= 85.7%,精度为87.1%,回忆= =每组的87.1%,F1得分= 87.0%)(图 3C)。一个工作流程,显示了实时卵母细胞准确性测试的过程以及如何使用Fluo-as-cast-bright管道选择NSN或SN卵母细胞进行潜在研究或或临床应用。 3D和E.
识别成像功能对于深度学习管道至关重要
在图2中的预测荧光图和明亮场图像之间的比较 4a,揭示了荧光预测网络学会了通过轻松地检测出有关核仁在明亮场图像中的数量和位置的明显信息来鉴定染色质结构的(图) 4a星号),以及相邻白染色质和异染色质区域的隐藏特征。重要的是,将预测的荧光图与固定和活卵母细胞的地面真相荧光图进行比较,揭示了显着的相似性,可以区分NSN卵母细胞中构素降解的程度差异,并忠实地代表了某些卵母细胞中两个核细胞中两个核的存在的差异。一个 4a,星号)。来自固定SN卵母细胞的预测荧光图表明,该网络还能够检测出明显的异染色质焦点或铬含量(箭头)和图像的大规模染色质构型模式,同时还放置了更明亮的鲜明和更多不同的染色质环的周围环境环。核仁。重要的是,固定NSN卵母细胞的预测荧光图在显示出占据整个生发囊泡的更反应的圣染色质结构方面也是准确的。另一方面,活的SN卵母细胞产生了昏暗的铬铬酸(图 4a;与从固定的SN卵母细胞(箭头)获得的预测的荧光图相比,所得的预测荧光图中的虚线矩形)主要是由于活卵母细胞中细胞内运动的增加。

一个真实和预测的荧光图之间的比较表明,荧光预测网络能够准确地识别核仁结构(星号)和关键的异染色质结构域(箭头和虚拟盒)。b类激活图说明了分类网络使用的图像中区域的重要性排名(颜色尺度)。
一些卵母细胞表现出短暂的,中间的染色质构型,其中,即使GV中的舌染色质仍被解剖并占据了生发囊泡内的大部分体积,围核酸的边缘也会在不同的程度上形成不同的程度(补充图。 3)。由于这些卵母细胞尚未完成过渡为SN配置,因此可能具有较低的发育潜力,因此我们的分类网络经过训练,可以将这个短暂的阶段指定为NSN,因为这些卵母细胞仍需要进行大量的大规模染色质重塑以达到SN配置并获得全部减数分裂和发育潜力。值得注意的是,由活的NSN卵母细胞产生的预测荧光图表现出反感的肠染色质,可以在分类步骤中准确鉴定(图。 4a)。
为了确定哪些功能对于分类网络最重要,我们使用了消融类激活图方法20,,,,21这显示了荧光图像中对于准确分类至关重要的区域。为了在用于分类的模型的输入图像中找到区域20,,,,21。该策略确定图像特征对于通过随机去除特征和对产生效果的比较而进行准确分类至关重要20,,,,21。从我们的所有三个分类模型中提取了激活图,并将平均类激活图绘制到用于分类的预测荧光图上。激活热图显示了在固定和活卵母细胞中染色质构型分类非常重要的区域。 4b。激活图表明,分类网络专注于不同卵母细胞中不同核区域以产生分类评分,主要集中在构素蛋白(EUC)以及突出的异染色质结构域上(图图。 4b;箭头),这是每个卵母细胞的染色质构型。重要的是,最关键的区域始终包括每个卵母细胞上的全部或部分染色质环或核圈,表明核仁周围的染色质环的亮度在分类中起着重要作用。
DNA小凹槽结合配体对核仁结构和卵母细胞的影响
将活卵母细胞暴露于可渗透的DNA结合配体污渍中,例如Hoechst 33342和核仁荧光标记物的显微注射,例如纤维蛋白-GFP,最近已用于识别显示的减数分裂作用的小鼠卵母细胞22,,,,23。然而,我们的结果表明,即使在短期暴露(25分钟)的活卵母细胞(25分钟)上,至0.5âg/ml Hoechst 33342,核仁结构也会严重损害(图。 5a和b)。在活卵母细胞的明亮场成像下检测到的核仁破坏的迹象包括显着增加(p= 0.0463)在表现出严重的核仁收缩和变形以及核仁片段的比例(56.6±11.5)中(43.3±15.3;p与对照组相比,= 0.0369)(图 5a和b)。值得注意的是,染色质结构的相应荧光染色显示核仁内部的弥漫性Hoechst染色和染色质片段的形成(图。 5b;箭头)。这些结果表明,即使短暂暴露于DNA结合配体(Hoechst 33342)也可以诱导暗示着核仁应力和活卵母细胞中碎屑形成的变化。

一个通过未处理的对照和25分钟的Hoechst处理,通过明亮场分析检测到具有核仁异常的百分比。误差线代表样本标准偏差(n= 10个卵母细胞的3个重复用于对照和治疗)。b卵母细胞核仁(*)的代表性明亮场图像及其相应的DAPI染色显示了Hoechst处理组中的核仁收缩和碎片。c在对照组和HOECHST治疗后的第一个小时(1 -H)或整个IVM(17 H)的HOECHST治疗中,体外成熟(IVM)的卵母细胞的百分比显示出染色体未对准或纺锤体异常(IVM)。误差线代表样本标准偏差(n= 10个卵母细胞的3个重复,用于对照和每种处理)。dHoechst暴露后,对照卵母细胞以及具有染色体错位或主轴和染色体异常的卵母细胞的代表性荧光显微照片。染色体显示为灰色,纺锤微管(乙酰化的α± - 微管蛋白)标记为绿色,并用红色描述了纺锤极Amtocs(塞酸酯)。星号表示统计学上的显着差异(p<0.05)。
为了确定活卵母细胞是否在体外成熟期间影响DNA结合配体会影响染色体分离(IVM),我们将卵母细胞暴露于0.5âg/ml HOECHST 33342在生发囊阶段或期间1 h hoechst 33342整个减数分裂成熟期(17 H)。在两个时间间隔内暴露于HOECHST 33342都会引起染色体隔离模式的大量异常(图。 5C和d)。统计分析表明,在两个HOECHST治疗组中,染色体未对准缺陷的卵母细胞的百分比明显更高(对照与HOECHST 1 H(51.3 -3-31.3);p= 0.034,对照与Hoechst 17 h(90±10);p= 0.037)。此外,在减数分裂成熟期间接触Hoechst 17小时导致显着增加(p = 0.046) in the proportion of oocytes (76.6 ± 11.5) that exhibited spindle structural abnormalities including unfocused and multipolar spindles, while chromosome structural abnormalities such as clustered chromosomes or scattered chromosomes also showed a significant increase (p = 0.046) after long term Hoechst exposure. These results indicate that even short-term exposure of live oocytes to Hoechst 33342 at the germinal vesicle stage may induce chromosome alignment defects and that long-term exposure during oocyte maturation exerts a highly detrimental effect on chromosome segregation and meiotic spindle stability.To determine whether exposure to Hoechst 33342 or microinjection of GFP-labeled nucleolar markers affects the patterns of gene expression in mouse oocytes, we compared two publicly available transcriptome data sets22,,,,
23that used a 15-min exposure to Hoechst 33342 or live cell imaging of Fibrillarin-GFP to select for NSN or SN oocytes22,,,,23。We found significant differences in the transcriptional profile of NSN oocytes selected by these two approaches (Fig. 6a)。For example, although 40% of transcripts exhibited similar levels of expression and read counts did not change between the Hoechst-treated group and the GFP-Fibrillarin microinjected group, 26% of transcripts exhibited a lower (absolute fold change ≥ 2) expression level and 34% of transcripts exhibited a higher (absolute fold change ≥ 2) expression level in the Hoechst treated group (Fig. 6a)。Differences in the transcriptional profile of SN oocytes selected by these two methods were also detected with 47% of transcripts showing higher expression levels and 20% of transcripts exhibiting lower expression in the Hoechst treated group (Fig. 6b)。Notably, the principal component analysis indicated that oocytes tend to cluster by method of selection rather than the type of chromatin configuration (Fig. 6C)。Gene ontology (GO) analysis revealed that the top 15 enriched terms for genes that showed significantly different read counts in the NSN oocytes correspond to factors involved in translation factor activity and RNA binding, histone binding, mRNA binding and histone deacetylase binding (Fig. 6d)。Moreover, the top 15 enriched GO terms for genes that show significantly different read counts in SN oocytes between the two approaches correspond to factors involved in ribonucleoprotein complex biogenesis and assembly, cell cycle, DNA replication, nuclear division, and ribosome biogenesis (Fig. 6E)。Examples of the log2 transformed gene expression levels of transcripts that showed significantly different read counts are represented in a heat map with their corresponding GO terms (Fig. 6f)。Genes related to nuclear functions (histone binding, nuclear division, DNA replication) and chromatin remodeling (deacetylase and kinase functions) show significantly different read counts.Importantly, we found significant changes in transcripts involved in ribosome metabolism, the chromatin reader LRWD1, and the Polycomb protein MBTD1 in Hoechst-treated oocytes as well as a significant 41.1-fold increase in transcript levels of the heat shock protein HSP90AA1 after Fibrillarin-GFP labeling (Fig. 6f)。These results indicate that even short-term exposure to Hoechst 33342 may significantly affect the patterns of expression of critical chromatin remodeling and ribosome biogenesis factors at the GV stage.Importantly, the increased levels of expression of heat shock protein HSP90AA1 after live cell imaging may suggest a potential response to genotoxic stress following exposure to ultraviolet light during prolonged fluorescence microscopy.Thus, these methods of separation between NSN and SN oocytes have a significant effect on the transcriptional profile of oocytes that may limit subsequent downstream applications.

一个和bComparison of publicly available transcriptome profiles22,,,,23。The percentage of genes that showed no significant change or significantly increased, or decreased expression (absolute fold change ≥ 2.0) in Hoechst-treated NSN or SN oocytes.cLow-dimensional representation of sample clustering generated from principal component analysis of the read counts.dTop 15 enriched GO terms for genes, which showed significantly different read counts in the NSN oocytes between the two studies (unadjustedp-values were used).eTop 15 enriched GO terms for genes, which showed significantly different read counts in the SN oocytes between the two studies (unadjustedp-values were used).fLog2-transformed gene expression levels of genes, which showed significantly different read counts represented in a heat map with their corresponding GO terms.
Transcriptome analysis following non-invasive selection of live oocytes with high developmental potential
Using our Fluo-Cast-Bright pipeline for the non-invasive selection of live NSN and SN oocytes, we analyzed and compared the transcriptome profile of these two types of oocytes using RNA sequencing (Fig. 7)。Fully-grown oocytes were obtained from the large antral follicles of non-superovulated female mice on day 20 of post-natal development.Single, denuded oocytes at the germinal vesicle stage were transferred to individual wells containing 100 μl MEM medium supplemented with 10 μM Milrinone and imaged under bright-field confocal microscopy (Fig. 7a)。The 3-D images of all collected oocytes were then processed immediately for nuclear segmentation and computational prediction of chromatin configuration.Live, GV-stage oocytes were then pooled based on the prediction of the Fluo-Cast-Bright pipeline into NSN (n = 15) and SN (n = 15) oocytes and processed for RNA sequencing in two replicates (Fig. 7a)。Fig. 7: Transcriptome profiles of live NSN and SN oocytes following non-invasive selection using Fluo-Cast-Bright.一个Experimental design: Live GV stage oocytes were individually imaged by bright-field microscopy, and 3-D image stacks were subjected to fluorescence prediction followed by classification using the Fluo-Cast-Bright pipeline as either NSN or SN stage oocytes. Pools of live oocytes (

 = 15) of each chromatin configuration were then subjected to RNAseq in two replicates.bPearson correlation plot.相关性 (r) values between the read counts of transcripts in each sample. Color scale represents the range of the correlation coefficients (r) displayed.cHeatmap of differentially expressed transcripts. Color scale indicates log2 transformed gene expression levels revealing clustering of samples by chromatin state.dLow-dimensional representation of sample clustering generated from principal component analysis based on the expression levels of 48574 mouse transcripts with not-null expression.eVolcano plot indicating differentially expressed transcripts. Significantly up- (red) or down- (green) regulated transcripts in SN oocytes are shown (问value < 0.05, absolute fold change > 2).Analysis of quality control parameters from our RNA-seq libraries such as total clean reads and total mapping ratio revealed a high reproducibility between experimental replicates (Supplementary Fig. 4)。
The Pearson’s correlation coefficient for global gene expression levels between every two samples (Fig. 7b) indicates that overall, the transcriptomes of NSN oocytes and SN oocytes are similar. However, analysis of differentially expressed transcripts detected a significant difference in expression levels for a subset of 707 transcripts between NSN and SN oocytes (Fig. 7C)。Importantly, dendrogram analysis of transcriptional profiles indicated that oocytes cluster by the state of chromatin configuration suggesting that significant differences can be detected in the transcriptional profile between NSN and SN oocytes (Fig. 7C)。Principal component analysis (PCA) using the “compressed variance†of differentially expressed transcripts clearly separated oocytes by chromatin state based on PC1 (Fig. 7d)。Notably, analysis of absolute loading values (Supplementary Fig. 5a) revealed that PC1 variance is accounted for by a cluster of highly abundant maternal transcripts, such as S-phase kinase-I (SKIP-1) a meiosis-specific subunit of the SCF (Skp1-Cullin1-F-box) complex required for meiotic competence24as well as members of the maternal subcortical complex such as NLRP14, ZBED3, OOEP and PADI6, which are required for female fertility25,,,,26,,,,27,,,,28。Gene ontology analysis revealed that these transcripts are involved in the regulation of protein metabolism and chromosome condensation (Supplementary Fig. 5b)。Comparison of differentially expressed transcripts between NSN and SN oocytes revealed that meiotically competent SN oocytes exhibit a significant (
问-value < 0.05), up regulation (absolute fold change > 2) of 365 transcripts and a significant down regulation of 342 transcripts (Fig. 7e)。Notably, the top-most up-regulated transcript in SN oocytes,Xpo1(exportin 1) exhibited a log2-fold overexpression of 24.5 (问-value = 6.37E−5). XPO1 is a nuclear transport receptor, also known as CRM1, that is involved in protein and RNA export from the nucleus, required for the transport of hundreds of proteins in both somatic cells and oocytes, and directly involved in oocyte GV breakdown and meiotic resumption29。Additional transcripts with similarly high fold up-regulation in SN oocytes were弗林(log2-fold change 22.8,问-value = 3.12E−4),Kif23(kinesin family member 23, also known as MKlp1; log2-fold change 22.8,问-value = 2.94E−4) andTbl1xr1(log2-fold change 23.1,问-value = 2.36E−04). Furin is a critical serine protease that cleaves immature pro-proteins into their corresponding active forms and regulates oocyte meiosis via Akt phosphorylation30。KIF23 is a microtubule kinesin involved in spindle assembly and remodeling31, while TBL1XR1 is a histone-binding component of the N-CoR and HDAC3 complex, which, exhibits chromatin-remodeling activity32。Other significantly up-regulated transcripts include the actin cytoskeleton regulatory factor33CAP1 (cyclase-associated actin cytoskeleton regulatory protein 1; log2-fold change = 10.1,问-value = 2.03E−7), which is involved in spindle migration, cortical actin cap formation and cytokinesis34, and transcripts forCnot6l(CCR4-NOT transcription complex subunit 6 like; log2-fold change = 9.88,问-value = 7.80E−6), which is involved in the degradation of maternal transcripts during oocyte maturation35,,,,36。Notably, the transcript with the most highly significant问-value in SN oocytes wasGpr1(G protein-coupled receptor 1;问-value = 2.39E−40), a gene that exhibits dynamic maternal imprinting in pre-implantation embryos37。
Amongst down-regulated transcripts in SN oocytes, we detectedNek1(NIMA-related kinase 1),Bcat1(branched-chain amino acid transaminase 1),Cnot3(CCR4-NOT transcription complex subunit 3), andGtse1(G2 and S-phase expressed 1). NEK1 (log2-fold change −23.7,问-value = 1.36E−4) is a hub signaling kinase that modulates DNA repair capacity and is involved in sensing and repair of DNA double-strand breaks at the G2-M transition in somatic cells38。The metabolic enzyme BCAT1 exhibited a log2-fold reduction of −24.5 (问-value = 6.95E−5). Bcat1 redox function is critical for the regulation of protein oxidation during mitosis and to maintain the centromeric localization of Aurora Kinase B39, while CNOT3 (log2-fold change = −23.3,问-value = 1.90E−4) is involved in the regulation of cell cycle progression40。GTSE1, the transcript that exhibited the most significant q value (log2-fold change = −1.40,问-value = 2.22E−21), is expressed exclusively during the late G2/M transition where it is known to play a role in the maintenance of G2 arrest and as a regulator of microtubule dynamics during mitosis41,,,,42。反应组43
enrichment analysis revealed that transcripts that are significantly up-regulated in SN oocytes are enriched for functional terms such as the formation of WDR5-containing histone-modifying complexes and epigenetic regulation of gene expression, the RHO GTPase, RAC1 GTPase, RHOA GTPase, and CDC42 GTPase cycles, which are essential for accurate completion of the metaphase-I to metaphase-II transition and cytokinesis44as well as deadenylation-dependent mRNA decay (Fig. 8a)。Whereas gene ontology (GO)45molecular function (MF) analysis revealed enrichment for factors associated with nucleoside-triphosphatase regulator activity, GTPase and transcription co-regulator activity (Fig. 8b)。Functional enrichment analysis of biological process (BP) revealed association with peptidyl-lysine modification and GTPase signaling, methylation and histone modification as well as biological processes related to chromosome segregation and chromatin remodeling (Fig. 8C)。Cellular component (CC) term enrichment analysis also revealed significant enrichment for factors related to the actin cytoskeleton, ribonucleoprotein granules and microtubules as well as histone deacetylase complex and the histone methyl transferase complex (Fig. 8d)。In contrast, we observed a down-regulation of factors associated with SUMOylation of DNA damage response and repair proteins in SN oocytes (Supplementary Fig. 6)。These results provide critical evidence indicating that cellular processes such as maternal mRNA deadenylation, histone post-translational modifications, epigenetic regulation of chromatin remodeling and signaling pathways for the control microtubule dynamics during the metaphase-I to metaphase-II transition are important components of the SN oocyte’s program for the acquisition of meiotic potential.

一个Enrichment of biological pathways based on Reactome database.bOverrepresented GO-Terms molecular function (GO_MF),c(biological process, GO_BP), anddcellular component (GO_CC). Shading indicates the level of significance, and the size of the bubble correlates with the number of transcripts per category (unadjustedp-values were used).
讨论
Non-invasive selection of mammalian oocytes with high meiotic and developmental potential is a pressing need in the fields of human infertility as well as animal reproduction. However, until now, the identification of reliable cellular and or molecular markers of oocyte quality and developmental potential that are compatible with cell viability, as well as downstream research or clinical applications has remained elusive. Here, we developedfluorescence prediction andcl作为sification on明亮的-field (Fluo-Cast-Bright), a novel pipeline for the non-invasive identification of chromatin structure and developmental potential in live mouse oocytes. Our convolutional neural network is capable of extracting information on the type of chromatin configuration present in fully-grown oocytes from non-invasive bright-field images obtained from live oocytes, generating a predicted fluorescent map of DAPI staining as well as H3K9me3 localization in the germinal vesicle. Our classification network was capable of distinguishing between oocytes that exhibit an NSN or SN configuration and thereby efficiently identified SN oocytes with high developmental potential. Fluo-Cast-Bright is thus capable to non-invasively and efficiently ‘cast’ a fluorescence prediction of large-scale chromatin structure on a bright-field image of unlabeled oocytes and achieved 91.3% accuracy in the classification of chromatin state in fixed oocytes and 85.7% accuracy in live oocytes. Importantly, our approach does not require any special imaging conditions or prolonged live cell imaging that may compromise cell viability, making it widely applicable in any conventional clinical setting or research laboratory.
As a proof of concept, we conducted the first transcriptome analysis following the non-invasive selection of live NSN and SN oocytes. Our results indicate that meiotically competent SN oocytes exhibit a higher expression of transcripts encoding for critical factors such as Exportin 1 (Xpo1), which is required for nuclear export of mRNA as well as hundreds of proteins during the G2/M transition, in addition to factors required for degradation of maternal mRNA stores, histone methylation as well as protein enzymatic activation. Importantly, our results indicate that many of these factors are required for the regulation of nuclear architecture, chromosome stability, the metaphase-I to metaphase-II transition, and the control of microtubule dynamics in preparation for the completion of meiosis. Our results reveal critical molecular markers for oocyte quality and provide additional evidence to dissect the molecular mechanisms regulating the oocyte’s program for nuclear and cytoplasmic maturation during the acquisition of meiotic and developmental potential. Importantly, our pipeline provides a fast and non-invasive selection of meiotically competent oocytes for downstream research and clinical applications.
Convolutional neural networks are powerful tools to derive quantitative subcellular information from bright-field or differential interference contrast microscopy images from somatic cells and tissues12,,,,13。Ours is the first pipeline specifically designed for the non-invasive analysis of nuclear architecture in live mammalian oocytes.A large body of evidence indicates that mouse SN oocytes exhibit higher meiotic and developmental potential1,,,,6,,,,9,,,,10,,,,11。For example, SN oocytes exhibit higher rates of meiotic maturation in vitro and development to the blastocyst stage following in vitro fertilization1,,,,6,,,,9,,,,10,,,,11。Thus, the use of our Fluo-Cast-Bright pipeline for the non-invasive identification of SN oocytes also allows for the efficient selection and sorting of live SN oocytes with high developmental potential.Our pipeline is capable of generating a prediction of chromatin state from a single-time-point, bright-field 3-D image of a live mouse oocyte taken within an average of 10 min under a conventional confocal microscope plus an additional 5 min for nuclear segmentation and computational image analysis.It achieved high accuracy NSN/SN classification performance on both fixed and live cells and generated fluorescence prediction maps that were remarkably similar to the corresponding ground truth fluorescent maps observed in fixed oocytes.Importantly, this prediction accuracy was obtained using 138 fixed and 105 live oocytes for training and validation, with tests demonstrating a robust and high performance of our model in all metrics evaluated here (accuracy, precision, recall, and F1 score).Our current studies are aimed at establishing whether these parameters can be further improved with the use of larger live cell imaging training data sets.We have recently demonstrated that the chromocenters within the SN oocyte nucleus exhibit high mobility in live oocytes8。This is consistent with the reduced definition of large pericentric heterochromatin domains (chromocenters) in the predicted fluorescent maps obtained following live oocyte imaging.However, re-training the classification networks using live oocytes was sufficient to recover the performance of our network and generate predicted fluorescent maps based on both readily apparent as well as hidden information on bright-field images that faithfully reflected the position of chromocenters in live oocytes。Thus, retraining the classification model alone was sufficient to recover the prediction accuracy and enabled us to determine the chromatin configuration of each live oocyte label-free.It is also possible that different laboratories may have different microscopes and/or objectives with different numerical apertures that yield bright field images with a different level of resolution.In which case, high-accuracy predictions will require a simple retraining step of the classification models to adjust for their image resolution levels.
Importantly, our pipeline provides a reliable indication of the chromatin state of live oocytes without affecting cellular viability, so that researchers or clinicians can make informed decisions while selecting oocytes for experiments or clinical applications.Fluo-Cast-Bright is also fast in generating a final chromatin prediction after nuclear segmentation, as our results indicate that batch prediction alone for 3-D image stacks of 42 oocytes takes only about 1.5 min on an Nvidia Tesla P100 graphics processing单元。
Several deep learning tools have been recently generated for the analysis of male and female gametes, albeit with different objectives. For example, a deep convolutional neural network was developed and trained using bright-field images of human spermatozoa with known DNA integrity to predict DNA quality in test samples, achieving 86th percentile in the prediction of DNA fragmentation index on sperm cells15。However, this network was developed for the analysis of fixed spermatozoa, and the algorithm will likely need drastic modifications and optimization procedures to work well on live cells as spermatozoa exhibit high motility and vigorous multidirectional movement.In another study, time-lapse bright field microscopy was used in combination with particle image velocimetry and a feed-forward artificial neural network for quantitative analysis of cytoplasmic movement velocity as a predictor of developmental competence in mouse oocytes16。However, this method requires a specialized cell-screening system, and the oocytes need to be imaged for 15 h, which limits the applicability of the protocol.More recently, oocyte phenotyping based on analysis of the texture of the zona pellucida and cytoplasmic particle size has shown some predictive value on oocyte maturation potential and to distinguish knockout oocytes with different genetic backgrounds46。Consistent with previous reports47this approach confirmed that small oocytes with a thin zona pellucida fail to undergo germinal vesicle breakdown and remain arrested at the GV stage. However, this approach does not distinguish between NSN oocytes, which are capable of meiotic maturation but have low developmental potential, and SN oocytes, which exhibit a higher potential for development to the blastocyst stage.
Both, DNA-binding ligands such as Hoechst 33342 or microinjection of a GFP-Fibrillarin marker have been recently used to separate mouse NSN and SN oocytes for experimental analysis22,,,,23。However, a direct comparison of transcriptional profiles in NSN and SN oocytes obtained using these two approaches suggests the presence of cytotoxic effects and DNA damage on these oocytes.Our results also indicate that even a brief exposure (25 min) to 0.5 μg/ml Hoechst 33342 induces striking changes in the nucleolar structure and chromatin configuration of oocytes.The nucleolus is a critical nuclear compartment for ribosome biogenesis, its morphology is known to correlate with the levels of rRNA transcription, cell growth and metabolic rates48。It also acts as a key hub in the cellular stress response.Thus, it is conceivable that the abnormalities we observe here originated from a stress response to DNA damage, which involves the activation of signaling pathways, that affect cell-cycle progression and trigger cellular senescence48。Consistent with this notion, oocytes microinjected with GFP-Fibrillarin23exhibit high levels of γ-H2AX phosphorylation, a bona fide marker for the presence of DNA damage and double-strand DNA breaks. The nucleolus is also an important platform for the organization of chromatin structure and regulation of genome integrity, thus the chromatin structural abnormalities we detected here after Hoechst exposure may have originated not only from DNA damage but also due to a disruption of the balance between ribosome biogenesis and chromatin organization49,,,,50。Importantly, in silico analysis and principal component analysis also revealed that this short exposure to Hoechst resulted in dramatic changes in gene expression involved in ribosome biogenesis consistent with the changes in nucleolar structure and chromosome instability.Many genes related to meiotic progression were also found to be differentially expressed between oocytes labeled by Hoechst22and oocytes labeled by fibrillarin-GFP23。Moreover, we found changes in the levels of expression for transcripts related to nuclear functions such as histone binding, cell division, and chromatin remodeling potentially accounting for the significant increase in chromosome and spindle abnormalities observed in Hoechst treated oocytes in this study.
To demonstrate downstream applications of our pipeline, we conducted a transcriptome analysis following a non-invasive selection of NSN and SN oocytes. This is the first time that an NSN and SN transcriptomics dataset has been generated without using any fluorescent labels and is, therefore a, faithful representation of transcriptomic changes in live oocytes during the NSN to SN transition. RNA-sequencing analysis revealed important transcriptional differences between these two types of oocytes. Both gene ontology (GO)45and Reactome43term analysis pointed to pathways related to chromatin, epigenetics, cell cycle regulation, and nuclear functions.
Notably, the most up-regulated transcript in SN oocytes encodes for the nuclear export receptor exportin 1 (Xpo1)。XPO1 is also known as CRM1 (chromosome region maintenance 1) and plays a critical role in the transport of myriad proteins51。Proteome analysis revealed close to 1000 proteins are CRM1 cargo substrates inXenopusoocytes, and their nuclear export is essential for critical cellular processes as diverse as centrosome function, cytoskeleton dynamics, ribosome maturation, and mRNA degradation52。Notably, chemical inhibition of XPO1 protein delays germinal vesicle breakdown, while over-expression accelerates this process in pig oocytes29。Thus, the accumulation of higherXpo1transcript and protein levels may be a critical step in preparation for meiosis onset and the acquisition of meiotic competence. This is consistent with previous studies, which revealed that NSN oocytes had a lower GVBD rate compared to SN oocytes23。Thus, our study provides evidence indicating thatXpo1transcript levels may be a critical molecular marker for oocyte quality and developmental potential.
Furin is a pro-protein convertase that is required for the proteolytic cleavage of numerous proteins into their active isoforms. Conditional deletion during mouse oocyte growth induces ovarian follicle arrest at the late pre-antral stage and results in subsequent infertility53。At the germinal vesicle stage, Furin is localized to the plasma membrane30, where it subsequently activates the Insulin-like growth factor 1 (IGF-1) receptor to maintain microvillus organization and activate protein kinase B (PKB/Akt) to promote the resumption of meiosis30,,,,54,,,,55。Our results indicate that SN oocytes also accumulate higher levels of transcripts required for spindle and actin filament regulation in preparation for meiotic progression, includingKif23和Cap1。KIF23/MKLP1 is recruited to the spindle mid zone by the inner centromere protein (INCENP) which is critical for mid-body formation56。In Drosophila oocytes, KIF23 exhibits a microtubule cross-linking kinesin activity that is essential for the regulation of meiotic spindle length and maintenance of microtubule stability during the metaphase-II stage31。During mouse oocyte meiosis, CAP1 co-localizes with the plasma membrane actin cap, where it is required for actin disassembly, spindle migration, asymmetric division, and polar body extrusion34,,,,57。Actin filament reorganization is essential for accurate chromosome segregation during meiosis I58, thus accumulation of higherCap1transcripts in SN oocytes may also contribute to the accurate segregation of chromosomes in oocytes of high developmental potential.
TBL1XR1 is an F-box-like/WD40 repeat-containing protein, and a member of the N-Cor complex32, known to functionally interact with Sycp3-like Y-linked (SLY), a multi-copy gene required for post-meiotic sperm chromatin remodeling59。尤其,Tbl1xr1is highly expressed as a maternal transcript in zebrafish embryos60。Maternal transcripts synthesized during oocyte growth undergo complex temporal regulation during meiotic maturation61。Selective degradation of maternal mRNA transcripts is essential for the acquisition of both meiotic and developmental competence.CNOT6L is a subunit of the CCR4–NOT complex, the eukaryotic mRNA deadenylase, that is preferentially expressed in mouse oocytes, its functional ablation interferes with mRNA degradation and results in meiotic spindle abnormalities, meiotic arrest, and severe subfertility36。Interestingly, the function of CNOT6L in maternal mRNA degradation is mediated through the RNA-binding protein ZFP36l2, which was recently shown to regulate global transcriptional silencing and chromatin modifications, including increased histone methylation (H3K9me3) associated with the transition into the SN configuration in mouse oocytes, providing a functional link between mRNA decay and global transcriptional quiescence during the acquisition of meiotic competence62。Thus, SN oocytes accumulate higher levels of key transcripts that are required not only for maternal mRNA degradation as well as large-scale chromatin remodeling but also higher levels of transcripts that are required for critical processes such as spindle assembly, migration, and remodeling to ensure accurate chromosome segregation during the metaphase-I to metaphase-II transition.
In contrast, we found that SN oocytes exhibit lower levels of transcripts for several critical factors associated with the response to DNA damage/repair compared with NSN oocytes. For example, NEK1 is critical for the detection of DNA double-strand breaks and its functional ablation in mitotic cells affects mitochondrial function, including a reduction in complex-I activity38。Notably, complex-I activity is known to be suppressed in a developmentally regulated manner in human andXenopusearly growing oocytes to prevent the formation of reactive oxygen species of mitochondrial origin63。BCAT1 is an essential metabolic enzyme that regulates branched-chain amino acid catabolism and is also required for the regulation of redox pathways39。Bcat1transcripts are similarly regulated and exhibit a conserved function during the process of physiological aging in秀丽隐杆线, zebra fish and mouse somatic cells64。Importantly, its induced overexpression has been recently associated with enhanced oocyte quality and extension of reproductive lifespan in the秀丽隐杆线模型65。Our results provide evidence that critical factors required for the repair of DNA damage and cellular aging are downregulated at the transcriptional level during the transition into the SN configuration, potentially reflecting a developmental switch in the DNA damage response and/or branched-chain amino acid catabolism associated with the increased chromatin condensation present in SN oocytes or perhaps as a result of the existence of specialized pathways to resolve DNA breaks during the G2/M transition in pre-ovulatory oocytes.Consistent with this notion, our recent studies revealed a striking difference in the chromatin response of NSN and SN oocytes to DNA double-strand breaks induced by acute γ-irradiation8。Our studies also provide molecular insight into the factors and potential mechanisms associated with the oocyte’s program for the acquisition of meiotic and developmental potential.Further studies will be required for the functional analysis of specific molecular markers associated with enhanced oocyte quality identified in this work.
In conclusion, our results provide evidence that deep learning networks can extract hidden information from bright-field images of germinal vesicle stage oocytes that normally only become visible with invasive labeling methods. Fluo-Cast Bright has potential for both research and clinical applications in human and animal reproduction. Similar strategies can be applied to other cell types, to facilitate a non-invasive analysis of cellular potency. For example, induced differentiation of cardiomyocyte or neuronal precursor cells from iPSC cells must retain high levels of DNA integrity in order to be considered for any potential therapeutic applications66。Thus, in silico prediction of DNA integrity and other cell quality attributes will be essential to expand the clinical applications of iPSC-derived cells with therapeutic potential.
方法
动物
All experiments were conducted in accordance with guidelines and following approval of protocols by the ‘Institutional Animal Care and Use Committee’ (IACUC) of the University of Georgia. We have complied with all relevant ethical regulations for animal use. Female mice of the speciesmus musculusand a hybrid genetic background of C57BL/6/DBA were housed at ambient temperature (24–26 °C) with a 12 h light/dark light cycle and food and water provided ad libitum and used for oocyte collection between postnatal days 18 and 22.
Oocyte collection and culture
GV stage oocytes were collected from non-primed female mice of a C57/Bl6/DBA2J F1 hybrid background between postnatal days 20–24 by follicular puncture. Cumulus oocyte complexes were then transferred to minimal essential medium (MEM), supplemented with 3 mg/ml bovine serum albumin (Sigma Aldrich, St. Louis, MO) and milrinone (Sigma; 1 μg/ml) to prevent meiotic resumption. Surrounding cumulus cells were removed by gentle pipetting and denuded oocytes were maintained at 37 °C in MEM/BSA plus milrinone under 5% CO2, 5% O2, and balanced N2until subjected to the procedures outlined below.Imaging of fixed oocytes following immunofluorescence stainingThe denuded oocytes were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) solution, supplemented with 0.05% Triton X-100 for 30 min at 37 °C and subsequently blocked in buffer containing 5% fetal bovine serum, antibiotic–antimycotic and 0.01% Triton X-100 in PBS at 4 °C. To reveal the state of chromatin in the nucleus, oocytes were immunolabeled with specific polyclonal antibodies directed against histone H3 trimethylated at lysine 9 (H3K9me3, 1:200, Cat#ab5819, Abcam, Cambridge, MA, USA) in blocking buffer overnight at 4 °C. Following repeated washes in blocking buffer, the cells were then incubated with a specific anti-rabbit Alexa Fluor 555-conjugated secondary antibody (1:1000, Cat#A21430, Life Technologies, Eugene, OR) for 1 h at 37 °C. After a series of final wash steps in blocking buffer, the oocytes in solution were exposed to DAPI (4’,6-diamidino-2-phenylindole) to counterstain the DNA and transferred to individual wells of optical bottom 96-well plates (Greiner Bio-One, Frickenhausen, Germany). Imaging was performed on a Nikon Eclipse Ti-U/D-Eclipse C1 laser-scanning confocal microscope equipped with a ×40 objective lens following sequential (frame lambda) excitation with 401 and 561 nm Coherent Sapphire lasers. Image acquisition was conducted using EZC1 3.91 software (Nikon) with a step size of 0.8 μm and a Z-stack range of ~12.8 μm to acquire 3-D image stacks of each oocyte with a bright-field channel, in addition to the fluorescence channels. The image stacks were taken at a resolution of 16*1024*1024 px
3
, with a pixel size of 800*148*148 nm3in the final 3D image stacks. The H3K9me3 and DAPI fluorescence signals were merged into a single chromatin fluorescence map. The cuboid volume in the image stack containing the nucleus was then segmented with the aid of labeling software labelme67and resized to 16*128*128 px3。The bright-field image and fluorescence map in these segmented image stacks were used as the training set.Each oocyte image stack was labeled as SN or NSN based on the combined fluorescence map of DAPI and H3K9me3.Imaging of live mouse oocytesFor live oocyte imaging, denuded oocytes were placed one by one in 180 µl micro-drops of MEM/BSA/milrinone on glass-bottom Petri dishes (Mattek, Ashland, MA) overlayed with mineral oil. The glass bottom dish was placed in an environmental chamber with an atmosphere of 5% CO
2
, 5% O2, and 90% N2at 37 °C. Imaging and segmentation were conducted exactly as described above. After imaging, the oocytes were then fixed and stained with DAPI to determine their true chromatin configuration.Hoechst treatment and in vitro maturation of GV oocytesTo observe the effect of DNA dyes on live GV oocytes, oocyte culture media was supplemented with 0.5 μg/ml of the minor groove DNA binding ligand Hoechst 33342 (Life Technologies, Eugene, OR) and oocytes were exposed for 20–25 min before imaging. The experiment was conducted in three biological replicates and compared to equal numbers of non-treated control oocytes (10 oocytes per group and replicate). Imaging and segmentation were conducted exactly as described above using a confocal microscope to obtain image stacks for fluorescence prediction as well as the detection of abnormalities caused by Hoechst treatment.
Control oocytes, as well as oocytes following short-term exposure (1 h) or continuous exposure (17 h) to 0.5 μg/ml Hoechst 33342 were subjected to in vitro maturation (IVM) for 17 h in DMEM supplemented with 5% FBS. For each control and treatment group, 3 replicates of 10 oocytes were used.
In vitro matured oocytes were then fixed, blocked, and immunostained as described above using a primary antibody cocktail consisting of a mouse anti-acetylated α-tubulin antibody (1:1000, T6793, Sigma Aldrich) and a rabbit anti-pericentrin antibody (1:1000, PRB432C, Covance, Princeton, NJ) in blocking buffer for 1 h at 37 °C. AlexaFluor-conjugated secondary antibodies included AF555-anti-rabbit and AF488-anti-mouse antibodies (1:1000, Life Technologies, Eugene, OR) After immunofluorescence labeling, the oocytes were mounted on glass slides using Vectashield mounting medium containing DAPI (VectaShield, Vector Laboratories, Burlingame, CA). The slides were then analyzed and imaged using a Leica DMRE epifluorescence microscope. Only oocytes with spindle orientations allowing unequivocal analysis were included in the data collection.
Comparison between two published oocyte transcriptome datasets
The transcriptome data analysis was carried out using the publicly available data sets comparing the transcriptomes of NSN and SN mouse oocytes, including Ma et al.
22
和Wang等。23。The average read counts of 757 genes common in both published datasets were used to carry out our analyses of the effect of Hoechst 33342 and Fibrillarin-GFP on transcriptome profiles, and genes with absolute fold change >2 were considered genes with a significantly different expression.First, we used the prcomp method in R
68to perform a principal component analysis on the read counts of 757 genes to determine the origin of variance in the two datasets (difference between SN vs. NSN or Hoechst vs. fibrillarin-GFP). Then, we compared the expression values of each gene in SN and NSN oocytes between the two studies. Read counts of the two datasets showing an absolute fold change ≥ 2 were considered differentially expressed in response to Hoechst exposure in SN as well as NSN oocytes. We performed functional annotation for each group by enrichment analysis based on the GO database45。fluorescence prediction and
cl作为sification onBrigHT-field (Fluo-Cast-Bright) pipeline Our Fluo-Cast-Bright pipeline involves a fluorescence prediction model followed by a classification step and is a software developed based on the pytorch framework 69。
The fluorescence prediction model is a CNN network with a 3D U-Net++18architecture (Supplementary Fig. 1a)。To train for fluorescence prediction, we applied modifications inspired by the pytorch-fnet of the Allen Institute for Cell Science13, such as training on batches of randomly sampled 3D patches of images. The network was also trained with a loss function, which guides the network to produce a fluorescence map with minimal mean-squared error to the actual combined fluorescence map of DAPI and H3K9me3.The training took place in the typical forward–backward fashion of CNNs for 300 epochs, with 50 steps in each epoch. The images fed into the network were 3-D image patches with a size of 16 × 64 × 64 px3
((ZXY) randomly sampled from the training set and stored in a patch buffer. The patch buffer started with 16 randomly sampled patches and replaced one of its patches with a newly sampled patch at an interval of about 31 steps. Training was performed with Adam optimizer and a learning rate of 0.001. The deep supervision function of the U-Net++ architecture was not applied.The classification network has the ResNeXt-10119
architecture (Supplementary Fig. 1B) and was trained to distinguish between SN and NSN oocytes using 2-D slices of 3D predicted fluorescence maps produced by the fluorescence prediction model. We used the predicted fluorescence map generated from validation images of the fluorescence prediction network to train the classification models, because of the resemblance of these predicted fluorescence maps to the fluorescence prediction network output from new oocyte images.For training, the 3-D predicted fluorescence maps were split into individual 2-D fluorescence maps, these 2-D fluorescence maps were shuffled and fed into the network in random batches with 32 images in each batch.Training was performed with a learning rate of 0.0005 on the Adam optimizer to maximize the accuracy of the NSN/SN classification.
The correct NSN/SN label of each 2-D fluorescence map was determined by the true NSN/SN chromatin state of each oocyte.The training was performed using three different train/valid splits to produce three different classification models.Training of a fluorescent prediction model takes ~65 min on the NVIDIA A100 Tensor Core graphic processing unit (300 epochs), while training of a classification model takes ~4 min on the NVIDIA A100 Tensor Core graphic processing unit (120 epochs)。
Training of the fluorescence prediction model was based on bright-field images of 96 3D fixed oocyte images as a training set and 42 3D fixed oocyte images as a validation set and was subsequently used for the fluorescence prediction of both fixed and live oocytes. In contrast, the training of the classification model for fixed oocytes was based on the 2D sections of 42 3D predicted fluorescence maps of fixed oocyte images, while training of the classification model for live oocytes was based on the 2D sections of 63 3D predicted fluorescence maps of live oocyte images. This difference in training between fixed and live oocyte images was deemed necessary to avoid decreased classification accuracy due to movements of subcellular structures and organelles in live but not in fixed oocytes, and the different classification models were subsequently applied according to oocytes that had not participated in training or validation to determine the chromatin configuration.
For the classification of the NSN or SN configuration using only bright-field images of oocytes that never participated in training or validation, a 3-D predicted fluorescence map was first produced with the fluorescence prediction network. Then the three classification networks scanned through each 2-D cross-section of the 3-D predicted fluorescence map to determine whether the fluorescence map reveals NSN or SN configuration. For each 2-D fluorescence map, each classification network provides a percentage score for NSN and SN. The average of the NSN and SN percentage scores predicted by the three models was then used to determine the label of the 2-D fluorescence map. The new live oocyte is then designated as NSN and SN based on the majority prediction of 2-D fluorescence maps.
We selected our model architectures based on the accuracy to predict chromatin configuration. The different parameters and optimal performance of our selected model architectures were examined by training and comparing eight commonly used architectures, as well as after conducting a parameter sweep (Supplementary Fig. 2A–D)。Three tests were carried out on each model architecture and each set of parameters.In each test, training was carried out using the same training and validation set images, and the best validation performance achieved by the model was recorded.We also tested the classification accuracy of eight different classification models by carrying out a prediction on the same set of new live oocyte images (Supplementary Fig. 2b)。
The publicly accessible ‘grad-cam’ tool20was used to carry out the ablation class activation map method21on our classification models to interpret how our pipeline reached its classification from predicted fluorescence maps. This method determines image features critical for accurate classification by stochastic removal of features and the resulting effect on output accuracy21(Fig. 4b)。
RNA-Seq on NSN and SN oocytes separated by the Fluo-Cast-Bright pipeline
We used our Fluo-Cast-Bright pipeline for the non-invasive selection of live oocytes with different chromatin state in two replicates. Oocytes were classified as either NSN or SN according to our pipeline. Oocytes with the NSN chromatin configuration were selected and pooled (n = 15), while the same number of oocytes with the SN configuration (n = 15) were selected and pooled. Oocytes of each chromatin state were processed for sample preparation, including removal of the zona pellucida using Tyrode’s solution and lysis for RNA-Seq70。Transcriptome analysis was carried out on a DNA nanoball sequencing platform, also known as the DNBSEQTM
平台。The genome reference we used for mapping is Genome Assembly GRCm38 (mm10).The data size generated from sequencing was about ~6 gb per sample and had an average genome mapping ratio of ~94.3% using HISAT2 (Version v2.0.4)71。The genome mapping ratios were very uniform, suggesting that the samples are comparable.Novel coding transcripts obtained from sequencing were first merged with reference transcripts to obtain the complete reference.Bowtie272(Version v2.2.5) was then used to map clean reads to this reference to calculate the gene expression level of each sample and achieved an average mapping ratio of ~82.9%.
Principal component analysis on the read counts of all 48,574 mouse transcripts with not-null expression was carried out using the PCA tool of Scikit-learn73。The feature importance of each transcript in PC1 was calculated as loadings, which represent the degree of association of features with the PC axis74。Loadings are calculated by the eigenvector coefficient of the feature in the PC multiplied by the square root of the eigenvalue (amount of variance summarized by PC)74。Transcripts with absolute loading greater than 1 were selected as transcripts of interest (Supplementary Fig. 5a) and GO term analysis was carried out on these transcripts (Supplementary Fig. 5b)。
Differential expression between NSN and SN samples was analyzed with DESeq275, and transcripts with an absolute fold change > 2 and问-value < 0.05 were used to identify differentially expressed genes (DEGs). Functional analyses were performed for these DEGs by enrichment analysis based on the GO database45and Reactome database43。
统计和可重复性
RNA-seq was conducted as a duplicate experiment, while the effect of Hoechst on oocyte morphology observations was carried out on three independent biological replicates. To ensure the stringency of comparison, the Pairwise Mann–Whitney你test (Wilcoxon signed-rank test) in R68was used to assess statistical significance. Data are presented as means, and variation between individual replicates is indicated as the standard deviations. The asterisk in bar plots represents a significance ofp-value < 0.05. Error bars on bar plots represent the sample standard deviation. GO term and Reactome enrichment analyses were carried out using the clusterProfiler76package in R.报告摘要
Further information on research design is available in theÂ
Nature Portfolio Reporting Summary链接到本文。数据可用性
Data generated during the current study have been deposited in NCBI GEO under accession number
GSE266845。代码可用性
Codes used for data analysis are available under a CC BY NC ND 4.0 (Creative Commons Attribution-Non-Commercial-No Derivatives 4.0 International License). ©2024 University of Georgia Research Foundation, Inc. (Fluo-Cast-Bright) was created by Zhang X, Baumann C, and De La Fuente R., at the University of Georgia (
https://github.com/XiangyuZhangCharlie/Fluo-Cast-Bright)。Zenodohttps://doi.org/10.5281/zenodo.14641311。参考De La Fuente, R. Chromatin modifications in the germinal vesicle (GV) of mammalian oocytes.
开发
生物。292 , 1–12 (2006).文章
一个 PubMed一个 Google Scholar一个 Ivanovska, I., Khandan, T., Ito, T. & Orr-Weaver, T. L. A histone code in meiosis: the histone kinase, NHK-1, is required for proper chromosomal architecture inDrosophila
oocytes.基因开发。19, 2571–2582 (2005). 文章一个
CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个 Zuccotti, M., Piccinelli, A., Rossi, P. G., Garagna, S. & Redi, C. A. Chromatin organization during mouse oocyte growth.摩尔。
复制。开发 41, 479–485 (1995).
文章一个 CAS一个 PubMed一个 Google Scholar一个
De La Fuente, R. & Eppig, J. J. Transcriptional activity of the mouse oocyte genome: companion granulosa cells modulate transcription and chromatin remodeling.开发生物。 229, 224–236 (2001).
文章一个 PubMed一个 Google Scholar一个
Miyara, F. et al. Chromatin configuration and transcriptional control in human and mouse oocytes.摩尔。复制。开发 64, 458–470 (2003).
文章一个 CAS一个 PubMed一个 Google Scholar一个
De La Fuente, R. et al. Major chromatin remodeling in the germinal vesicle (GV) of mammalian oocytes is dispensable for global transcriptional silencing but required for centromeric heterochromatin function.开发生物。 275, 447–458 (2004).
文章一个 PubMed一个 Google Scholar一个
Longo, F. et al. Nuclear localization of NORs and centromeres in mouse oocytes during folliculogenesis.摩尔。复制。开发 66, 279–290 (2003).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Baumann, C. et al. Acute irradiation induces a senescence-like chromatin structure in mammalian oocytes.社区。生物。 6, 1258 (2023).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Zuccotti, M., Garagna, S., Merico, V., Monti, M. & Redi, C. A. Chromatin organisation and nuclear architecture in growing mouse oocytes.摩尔。细胞。内分泌。 234, 11–17 (2005).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Zuccotti, M. et al. Maternal Oct-4 is a potential key regulator of the developmental competence of mouse oocytes.BMC开发。生物。 8, 1–14 (2008).
文章一个 Google Scholar一个
Monti, M. et al. Developmental arrest and mouse antral not-surrounded nucleolus Oocytes1.生物。复制。 88, 1–7 (2013).
文章一个 Google Scholar一个
Christiansen, E. M. et al. In silico labeling: predicting fluorescent labels in unlabeled images.细胞 173, 792–803 (2018).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Ounkomol, C., Seshamani, S., Maleckar, M. M., Collman, F. & Johnson, G. R. Label-free prediction of three-dimensional fluorescence images from transmitted-light microscopy.纳特。方法 15, 917–920 (2018).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Djeghloul, D. et al. Loss of H3K9 trimethylation alters chromosome compaction and transcription factor retention during mitosis.纳特。结构。摩尔。生物。 30, 489–501 (2023).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
McCallum, C. et al. Deep learning-based selection of human sperm with high DNA integrity.社区。生物。 2, 250 (2019).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Cavalera, F. et al. A neural network-based identification of developmentally competent or incompetent mouse fully-grown oocytes.J. Vis。经验。e56668https://doi.org/10.3791/56668(2018)。
Zhang, X., Baumann, C. & De La Fuente, R.Fluo-Cast-Bright https://github.com/XiangyuZhangCharlie/Fluo-Cast-Bright。https://doi.org/10.5281/zenodo.14641311(2024)。Zhou, Z., Siddiquee, M. M. R., Tajbakhsh, N. & Liang, J. Unet++: redesigning skip connections to exploit multiscale features in image segmentation.
IEEE Trans。医学成像 39, 1856–1867 (2019).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Xie, S., Girshick, R., Dollár, P., Tu, Z. & He, K. Aggregated residual transformations for deep neural networks.在Proc。IEEE Conference on Computer Vision & Pattern Recognition1492–1500 (2017).
Gildenblat, J. A. C.Advanced AI Explainability for PyTorch http://github.com/jacobgil/pytorch-grad-cam(2021)。
Desai, S. & Ramaswamy, H. G. Ablation-cam: visual explanations for deep convolutional network via gradient-free localization.在Proc。IEEE/CVF Winter Conference on Applications of Computer Vision983–991 (2020).
Ma, J.-Y.等。Maternal factors required for oocyte developmental competence in mice: transcriptome analysis of non-surrounded nucleolus (NSN) and surrounded nucleolus (SN) oocytes.细胞周期 12, 1928–1938 (2013).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Wang, T. & Na, J. Fibrillarin-GFP facilitates the identification of meiotic competent oocytes.正面。细胞开发。生物。 9, 648331 (2021).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Kinterová, V., Kaňka, J., Bartková, A. & Toralová, T. SCF Ligases and their functions in Oogenesis and embryogenesis—summary of the most important findings throughout the animal kingdom.细胞 11, 234 (2022).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Zhang, W. et al. NLRP14 deficiency causes female infertility with oocyte maturation defects and early embryonic arrest by impairing cytoplasmic UHRF1 abundance.细胞代表。 42,,,,https://doi.org/10.1016/j.celrep.2023.113531(2023)。
Gao, Z. et al. Zbed3 participates in the subcortical maternal complex and regulates the distribution of organelles.J. Mol。细胞生物。 10, 74–88 (2018).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Tong, X. et al. Mutations in OOEP and NLRP5 identified in infertile patients with early embryonic arrest.哼。变形。 43, 1909–1920 (2022).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Xu,Y。等。Mutations in PADI6 cause female infertility characterized by early embryonic arrest.是。J. Hum。基因。 99, 744–752 (2016).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Onuma, A., Fujioka, Y. A., Fujii, W., Sugiura, K. & Naito, K. Effects of exportin 1 on nuclear transport and meiotic resumption in porcine full-grown and growing oocytes.生物。复制。 98, 501–509 (2018).
文章一个 PubMed一个 Google Scholar一个
Shi, L.-Y.等。Placenta-specific 1 regulates oocyte meiosis and fertilization through furin.FASEB J. 32, 5483–5494 (2018).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Costa, M. F. & Ohkura, H. The molecular architecture of the meiotic spindle is remodeled during metaphase arrest in oocytes.J. Cell Biol。 218, 2854–2864 (2019).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Yoon, H.-G., Choi, Y., Cole, P. A. & Wong, J. Reading and function of a histone code involved in targeting corepressor complexes for repression.摩尔。细胞。生物。 25, 324–335 (2005).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Ono, S. The role of cyclase-associated protein in regulating actin filament dynamics—more than a monomer-sequestration factor.J. Cell Sci。 126, 3249–3258 (2013).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Jin, Z.-L., Jo, Y.-J., Namgoong, S. & Kim, N.-H. CAP1-mediated actin cycling via ADF/cofilin proteins is essential for asymmetric division in mouse oocytes.J. Cell Sci。 131, jcs222356 (2018).
文章一个 PubMed一个 Google Scholar一个
Vieux, K.-F. & Clarke, H. J. CNOT6 regulates a novel pattern of mRNA deadenylation during oocyte meiotic maturation.科学。代表。 8, 6812 (2018).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Sha, Q. Q. et al. CNOT 6L couples the selective degradation of maternal transcripts to meiotic cell cycle progression in mouse oocyte.Embo J. 37, e99333 (2018).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Duffié, R. et al. The Gpr1/Zdbf2 locus provides new paradigms for transient and dynamic genomic imprinting in mammals.基因开发。 28, 463–478 (2014).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Martins, M. B. et al. NEK1 deficiency affects mitochondrial functions and the transcriptome of key DNA repair pathways.Mutagenesis 36, 223–236 (2021).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Francois, L. et al. BCAT1 redox function maintains mitotic fidelity.细胞代表。 41,,,,https://doi.org/10.1016/j.celrep.2022.111524(2022)。
Takahashi, A. et al. Involvement of CNOT3 in mitotic progression through inhibition of MAD1 expression.生物化学。生物。res。社区。 419, 268–273 (2012).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Bendre, S. et al. GTSE1 tunes microtubule stability for chromosome alignment and segregation by inhibiting the microtubule depolymerase MCAK.J. Cell Biol。 215, 631–647 (2016).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Tipton, A. R., Wren, J. D., Daum, J. R., Siefert, J. C. & Gorbsky, G. J. GTSE1 regulates spindle microtubule dynamics to control Aurora B kinase and Kif4A chromokinesin on chromosome arms.J. Cell Biol。 216, 3117–3132 (2017).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Gillespie, M. et al. The reactome pathway knowledgebase 2022.Nucleic acids Res. 50, D687–D692 (2022).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Sharma, P. & Beato, M. Long non-coding RNAs are driver to maintain the chromatin active regions at divergent transcriptional units.Non-coding RNA Investig. 2,,,,https://doi.org/10.21037/ncri.2018(2018)。
Consortium, G. O. The Gene Ontology (GO) database and informatics resource.核酸res。 32, D258–D261 (2004).
文章一个 Google Scholar一个
Letort, G. et al. An interpretable and versatile machine learning approach for oocyte phenotyping.J. Cell Sci。 135, jcs260281 (2022).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Hirao, Y., Miyano, T. & Kato, S. Acquisition of maturational competence in in vitro grown mouse oocytes.J. Exp。zool。 267, 543–547 (1993).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Boulon, S., Westman, B. J., Hutten, S., Boisvert, F.-M. & Lamond, A. I. The nucleolus under stress.摩尔。细胞 40, 216–227 (2010).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Narla, A. & Ebert, B. L. Ribosomopathies: human disorders of ribosome dysfunction.血 115, 3196–3205 (2010).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Grummt, I. Wisely chosen paths–regulation of rRNA synthesis.FEBS J. 277, 4626–4639 (2010).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Fung, H. Y. J., Fu, S.-C., Brautigam, C. A. & Chook, Y. M. Structural determinants of nuclear export signal orientation in binding to exportin CRM1.Elife 4, e10034 (2015).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Kırlı, K. et al. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning.Elife 4, e11466 (2015).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Meng, T.-G.等。Oocyte-specific deletion of furin leads to female infertility by causing early secondary follicle arrest in mice.细胞死亡。 8, e2846–e2846 (2017).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Scamuffa, N. et al. Selective inhibition of proprotein convertases represses the metastatic potential of human colorectal tumor cells.J. Clin。调查。 118, 352–363 (2008).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Jaaks, P. et al. The proprotein convertase furin contributes to rhabdomyosarcoma malignancy by promoting vascularization, migration and invasion.PLOS一个 11, e0161396 (2016).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Zhu, C., Bossy-Wetzel, E. & Jiang, W. Recruitment of MKLP1 to the spindle midzone/midbody by INCENP is essential for midbody formation and completion of cytokinesis in human cells.生物化学。J. 389, 373–381 (2005).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
DesMarais, V., Macaluso, F., Condeelis, J. & Bailly, M. Synergistic interaction between the Arp2/3 complex and cofilin drives stimulated lamellipod extension.J. Cell Sci。 117, 3499–3510 (2004).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Mogessie, B. & Schuh, M. Actin protects mammalian eggs against chromosome segregation errors.科学 357, eaal1647 (2017).
文章一个 PubMed一个 Google Scholar一个
Moretti, C. et al. SLY regulates genes involved in chromatin remodeling and interacts with TBL1XR1 during sperm differentiation.细胞死亡不同。 24, 1029–1044 (2017).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Jia, Y., Jiang, Q. & Sun, S. Embryonic expression patterns of TBL1 family in zebrafish.基因Expr。模式 51, 119355 (2024).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Clarke, H. J. Post-transcriptional control of gene expression during mouse oogenesis.在Mouse Development: From Oocyte to Stem Cells(ed Kubiak, J. Z.) 1–21 (Springer, Berlin, Heidelberg, 2012).
Chousal, J. N. et al. Chromatin modification and global transcriptional silencing in the oocyte mediated by the mRNA decay activator ZFP36L2.开发细胞 44, 392–402.e397 (2018).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
RodrÃguez-Nuevo, A. et al. Oocytes maintain ROS-free mitochondrial metabolism by suppressing complex I.自然 607, 756–761 (2022).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Mansfeld, J. et al. Branched-chain amino acid catabolism is a conserved regulator of physiological ageing.纳特。社区。 6, 10043 (2015).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Lesnik, C. et al. Enhanced branched-chain amino acid metabolism improves age-related reproduction in秀丽隐杆线。纳特。分子。1–17https://doi.org/10.1038/s42255-024-00996-y(2024)。
Assou, S., Bouckenheimer, J. & De Vos, J. Concise review: assessing the genome integrity of human induced pluripotent stem cells: what quality control metrics?干细胞 36, 814–821 (2018).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Wada, K. Labelme: Image Polygonal Annotation with Pythonhttp://github.com/wkentaro/labelme(2020)。
Ihaka, R. & Gentleman, R. R: a language for data analysis and graphics.J. Comput。图形。统计 5, 299–314 (1996).
文章一个 Google Scholar一个
Paszke, A. et al. Pytorch: an imperative style, high-performance deep learning library.ADV。神经信息。过程。系统。 32, 721 (2019).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq.纳特。方法 5, 621–628 (2008).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype.纳特。生物技术。 37, 907–915 (2019).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Langdon, W. B. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks.BioData Min. 8, 1–7 (2015).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Pedregosa, F. et al. Scikit-learn: machine learning in Python.J. Mach。学习。res。 12, 2825–2830 (2011).
Peres-Neto, P. R., Jackson, D. A. & Somers, K. M. Giving meaningful interpretation to ordination axes: assessing loading significance in principal component analysis.生态 84, 2347–2363 (2003).
文章一个 Google Scholar一个
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.基因组生物。 15, 1–21 (2014).
文章一个 Google Scholar一个
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters.OMICS16, 284–287 (2012).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
致谢
This work was supported by research grants from the U.S. Department of Agriculture National Institute of Food and Agriculture-National Institutes of Health dual-purpose grant (2020-67105-30882) to R. De La Fuente. Research in the author’s laboratory is also funded by the National Science Foundation Center for Cell Manufacturing grant (CMaT EEC-1648035). In Memoriam: This work is dedicated as a humble tribute to the loving memory of Carmelita Lozada De La Fuente+。
道德声明
竞争利益
A patent application (No. 63/564,218) has been filed on behalf of the authors by the University of Georgia Research Foundation related to the work presented in this manuscript.
同行评审
同行评审信息
通信生物学thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Kaliya Georgieva.
附加信息
Publisher’s note关于已发表的地图和机构隶属关系中的管辖权主张,Springer自然仍然是中立的。
补充信息
权利和权限
开放访问本文已根据创意共享归因非商业 - 非涉及商业4.0国际许可证获得许可,该许可允许以任何媒介或格式的任何非商业用途,共享,分发和复制,只要您对原始作者提供适当的信誉来源,提供了与Creative Commons许可证的链接,并指示您是否修改了许可材料。您没有根据本许可证的许可来共享本文或部分内容的改编材料。The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.要查看此许可证的副本,请访问http://creativecommons.org/licenses/by-nc-nd/4.0/。重印和权限
引用本文

Zhang, X., Baumann, C. & De La Fuente, R. Fluo-Cast-Bright: a deep learning pipeline for the non-invasive prediction of chromatin structure and developmental potential in live oocytes.
社区生物8 , 141 (2025). https://doi.org/10.1038/s42003-025-07568-0下载引用
:2024年7月1日
:2025年1月17日
:2025年1月29日
:https://doi.org/10.1038/s42003-025-07568-0https://doi.org/10.1038/s42003-025-07568-0