
使用机器学习潜力在镁中扩散的氢扩散:比较研究
作者:Franchini, Cesare
介绍
可持续和绿色能源解决方案的全球必要性加剧了寻找有效的氢存储材料的搜索。燃料之间的能量密度最高1,氢代表可以用零CO生产的化石源的有希望的替代品2由剩余可再生能源提供动力的排放2,通过诸如电解等方法3,,,,4,,,,5。阻止基于氢能的未来经济的主要障碍是生产成本,并且没有绿色,安全和有效的方式来存储和运输它。在这方面,固态氢存储技术是最安全的,提供更高的体积密度6比低压或高压气态替代品7,,,,8,,,,9。尽管有这些优势,但该技术仍处于早期阶段,并寻找允许大规模应用的材料,例如在汽车行业中10,保持开放11,,,,12。
金属氢化物当前代表有希望的有效且经济的解决方案13。特别是,镁以其出色的氢存储能力而脱颖而出14,环境友好性和自然丰度,显示高达7.6%wt的理论存储能力15。但是,基于镁的化合物中氢的缓慢动力学仍然对可能的应用构成了限制。因此,通过理论建模理解和优化氢扩散途径16,,,,17,,,,18和实验研究19,,,,20,,,,21为了改善未来基于MG的氢存储材料的性能至关重要。尽管在过去十年中进行了许多努力,但建模固态化合物中的氢动力学仍然具有挑战性22,,,,23,,,,24,,,,25。镁的低氢扩散性需要按纳米秒的顺序进行延长的模拟时间,以便使用AB-Initio分子动力学(MD)进行准确的研究。因此,Ab-Initio过渡状态计算,例如轻度弹性频段(NEB)方法,已成为重现和解释迄今为止文献中记录的实验数据的最有效方法17。但是,这种技术很麻烦,对于具有较高缺陷浓度或复杂势能景观的系统通常不切实际:手动描述NEB所需的所有可能的路径可能非常具有挑战性,几乎是不可能的17。
最近,机器学习加速MD(MLMD)通过对长时间尺度上可访问的大型系统进行准确的模拟,彻底改变了MD的世界26。这种方法在研究氢缺陷系统中的应用引起了人们的兴趣27,由于对动态属性的预测将大大扩展当今的过渡状态计算提供的有限景观。MLMD确实允许研究多组分系统28并可以有效地说明缺陷之间的相互作用29。但是,众所周知25,,,,30。尽管如此,该领域仍在迅速增长,并提出了几种新方法来探索新的和复杂的相位空间。一方面,各种预训练的通用解决方案31,,,,32开始可用,旨在提供一种方便而多功能的解决问题的方式。但是,正如这项工作中讨论的那样,尽管他们的训练数据集富含化学成分空间,但有限的配置采样可以显着损害其在以前看不见的缺陷,可稳态和过渡状态上的准确性,从而导致未呈现的概括能力。另一方面,由于构建了直通数据库33,,,,34。以错误为导向的配置采样使这些模型可以轻松地收集高质量的数据,从而广泛跨越配置空间,从而使它们具有高度准确性,尽管它们的体系结构与神经网络相比受到限制。当前的研究旨在说明在扩散动态条件下用于ML电位应用的系统程序,该程序可用于增强不同嵌入式缺陷的研究,而无需脱离AB-Initio的准确性。该过程特别包括通过积极学习的配置来改善基于ML的预训练模型,该模型是通过维也纳AB Initib in Initib in Initib in Initibal仿真软件包贝叶斯ML力场(VASP-MLFF)生成的。33,,,,34。我们考虑四个不同的浓度(MGH0.03125,MGH0.046875,MGH0.0625和MGH0.078125)并采用适当的方法来确保对无偏的动力学特性进行准确分析,并在三个不同的温度(300 K,480 K和673 K)下计算氢扩散系数。两个不同的通用跨性别电势(UIP),CHGNET31和狼牙棒32,考虑了。与UIP的预训练和微调版本一起计算了VASP-MLFF的动力学特性。不同结果和实验数据之间的比较表明,与VASP-MLFF和微调电位相比,同时训练的版本无法达到令人满意的精度。有趣的是,通过正确预测扩散系数的温度依赖性,微调电位优于VASP-MLFF。
结果
验证ML电位
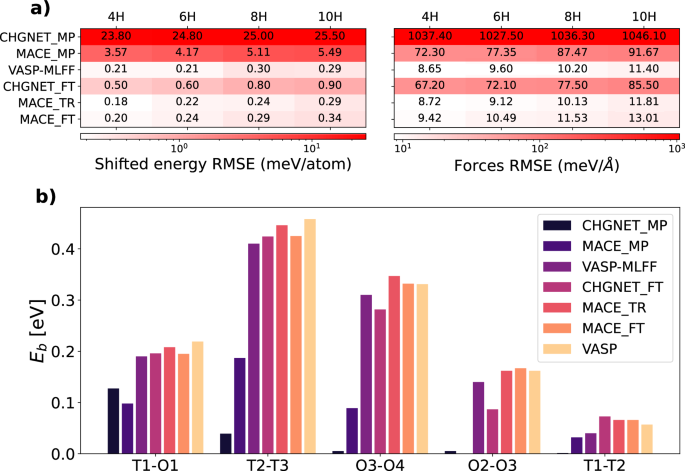
结果在图中报告。1显示VASP-MLFF的预测如何在0.3 MeV/Atom以下的精度下分别用于能量和10 MeV/ã的力量,而与其他涉及MLFF-MD的研究相比,相当预期35,,,,36,,,,37,,,,38。另一方面,在MP数据库中进行了预先训练的UIP,分别在CHGNET_MP和MACE_MP上命名,未能达到超过一个数量级的准确性水平。对VASP生成数据库的两个模型进行微调后,差异显着降低:获得的误差表明,CHGNET_FT性能达到的水平与VASP-MLFF相当,而MACE_FT均超过了它。CHGNET_FT和MACE_FT之间的性能差异可能源于后者所采用的模棱两可的架构。与CHGNET(例如CHGNET)相比,MACE的数据效率高度高,从小数据集获得了更好的微调结果。MACE模型还接受了VASP-DFT配置(MACE_TR)的训练,相对于MACE_FT,在力上实现了稍小的错误。请注意,与训练浓度不同的所有能量值都表现出取决于氢含量的系统转移。但是,氢动力学的潜在物理学并不受影响,因为在整个模拟过程中,这种转移保持恒定数量的颗粒数量,并且不会影响力。因此,模型仍然能够准确描述氢动力学。为了完整性,我们还介绍了原始验证和相应的r2值,如补充图所示。SF2。图1:不同ML电位的验证结果。一个

这些结果以不同的浓度给出,使用具有可变数量的H原子的超级细胞获得,从而通过将所有构型的平均值以相同的浓度设置为零来消除相应的恒定能移动。下标MP指的是材料项目数据库中的预培训模型,即从从头开始训练的一个训练的模型,然后to to to to-ft-ft to to-ft-untuned版本。b与不同八面体之间的氢过渡相关的能屏障值(O我)和四方(t我)位点,使用不同的ML电位和DFT参考(以黄色)获得通过CineB计算获得。用ML电位进行的攀爬图像弹性带(Cineb)计算,允许估计过渡能屏障eb
在各种八面体和四方氢间质位点之间,并进一步验证ML电位的准确性。实际上,我们比较了所获得的eb通过这些方法具有VASP-DFT基准测试结果的值。我们的发现,如图所示。1B,揭示了通用电位在预测能屏障时的表现明显精确。但是,受过训练和微调的对应物的性能会大大改善,从而产生更接近VASP-DFT参考值的结果。VASP-MLFF方法还与DFT基准相同。正如预期的那样,该模型在验证集上具有更好的精度,尤其是在力上,获得的结果更接近DFT值。
扩散系数和动态分析
扩散系数d使用“方法”部分中描述的方法为每个模型预测,如图2所示。2A,并与实验19和neb17结果,在每个研究的温度下。Nishimura等人的实验数据。收集在474â493k的温度范围内,然后使用Arrhenius关系在较高和较低的温度下推断。结果清楚地表明,我们的程序提供了具有出色实验协议的多个解决方案,超过了NEB计算16到目前为止,这已经代表了此类应用程序的标准。特别是对于MACE_FT的潜力而言,这是正确的,它不仅可以预测所有温度的正确数量级,而且在480 K. 480k。相反,VASP-MLFF在480 K和673 K处显示出良好的一致性,同时缺少一个数量级的室温。关于CHGNET_FT,它与480 K处的实验值相当一致,但是在最低温度下,低估了一个数量级的结果。正如对能量和力的误差分析所预期的那样,UIP的两个预训练的版本都会导致一致性较低,并且在大多数温度下,偏离至少一个数量级的偏差。如图2所示,仔细检查了扩散系数的温度依赖性。2B,揭示CHGNET_FT仍然通过更好地再现实验中观察到的ARRHENIUS曲线来优于VASP-MLFF,而MACE_FT Solutions均优于两者。这显示了如何使用较小的误差,就贝叶斯替代方案而言,深层网络能够更好地代表能量景观的形状。在这方面,可以通过比较激活能的预测值来提供进一步的定量分析。一个,从Arrhenius图中的线性拟合获得。特别是,我们实现了VASP-MLFF的0.4 eV,CHGNET_FT的0.19 eV和MACE_FT的0.28 eV,其中实验值对应于0.25 eV19。MACE_FT显然擅长代表系统的能量景观。图2:不同ML电位的模拟结果。一个

0.0625在三个不同的恒定平均温度下为380 k,480 k和673 k,以及相应的激活能,比较了我们的研究中通过不同的电位获得的结果,并且先前的研究17,,,,19。括号中的数字指示该值的最后一个重要数字中平均值(SEM)的标准误差。配色栏在logscale中突出显示相对于实验值的相对偏差。b对于ML模型,NEB的扩散系数与温度的依赖关系之间的比较17和实验结果19。实验曲线是从474 493 K.的数据中推断出来的。c在1 ns长NVE模拟中,原子对的径向分布函数。VASP-MLFF和MACE对整个域显示了与整个域的显着一致性,而CHGNET在较大的距离时开始有所不同。
此外,我们计算了d在各种氢浓度下使用MACE_FT以及相关的E一个,如图图所示。3一个。结果表明,随着浓度的增加,氢迁移率降低,导致扩散系数的相应降低。同时,随着较高的氢密度,活化能增加。评估d在不同的浓度中,可以与实验值进行比较,表明较低浓度的结果,特别是MGH0.03和MGH0.045,显示最好的对齐。应该指出的是Nishimura等。19未报告样品中的氢浓度。这些发现表明,Nishimura的实验中的靶氢浓度可能低于0.0625,在先前的DFT研究中假定17。

在不同温度和H浓度下的扩散系数在顶部,并在不同浓度的底部获得活化能。参考的目标实验值。19在每个图中报告为虚线。b比较模拟氢对分布(np在三个温度(300 k,480 k和673 k)下,出现理论泊松分布(点和虚线),用于不同的氢浓度。下面板显示我”参数(平均成对数)作为温度和浓度的函数。通过评估执行的所有NVE运行中的径向分布函数(RDF),对系统的动力学进行了进一步的分析。
我们在图中报告。2c系统中最佳性能模型的预测行为在673 k处,而其他温度案例可以在补充图中找到。SF6。发现MACE_FT和VASP-MLFF之间的一个很好的一致性,而CHGNET_FT的RDF通过平滑峰来远离其他距离。从这样的曲线可以保留有关模拟过程中氢行为的信息。在距离半径增加的情况下进行程序,第一个峰出现在Mg h曲线的2字前,与一个镁原子的平均距离距离最近的八面体位点的中心的平均距离对应16。属于H h对的第二个峰值在2.6大左右非常明显,反映了此距离处的氢原子之间的相关性。这种行为在文献中与氢化镁纳米簇的分子动力学一致30。实际上,2.6的距离对应于沿C方向的两个八面体位点之间的距离16,正如文献中报道的30,这意味着氢如何倾向于坐在附近的位置。为了强调这种行为,我们还报告了图。4在100 ps模拟期间,在673 K.时,镁基质内代表性氢原子的广泛扩散路径。该轨迹在周期性图像的复制品中解开,以增强可见性和解释。颜色梯度用作时间标记,蓝色指示氢原子在模拟开始时的初始位置(t= 0 ps)和红色指示其在研究间隔结束时的位置(t= 100 ps)。中间颜色(青色,绿色,黄色和橙色)代表这两个极端之间的时间发展,为原子扩散路径的时间演变提供了视觉提示。黑色圆圈突出了氢原子倾向于在镁位点振荡的间隙区域,表明在继续其扩散轨迹之前,表明晶格结构内的临时捕获位点。在Mg毫克曲线中观察到RDF中的第三个显着峰,与HCP结构中的典型镁距离保持一致。多个较小的峰表明晶格中的进一步邻居相互作用。在300 k和480 k的RDF中发现了类似的结果,由于热运动的效应降低,峰更加清晰,请参见补充图。SF6。特别是,在较低的温度下,较高的RDF表明氢倾向于在镁附近花费更多的时间,而在晶体结构中的扩散较少。

黑色圆圈突出了h原子在继续扩散轨迹之前倾向于在mg晶格位点振荡的间质区域。
氢对的分析(np图中显示的分布。3B对成对形成对温度和浓度的依赖性提供了清晰的见解。在升高的温度(例如480 k和673 k)时,模拟分布与理论泊松分布紧密一致,计算为:$ f({n} _ {p}; \ lambda)= \ frac {{e}^{ - \ lambda} {\ lambda} {\ lambda}^{n} {n} _ {p}}}}}}}}}}}}}}
(1)
在哪里
我”表示对的平均数量。该协议证明了氢配对的随机性质,表明与初始原子构型相关性缺乏相关性和独立性。此外,我”随着温度的增加,不同浓度的值强调了这种内在的随机性。相反,在300 k处,与泊松行为的偏差出现了,我”表现出对氢浓度的更强依赖性。这种行为可以归因于低温下氢原子的迁移率降低,在低温下,热能不足以克服间质位点过渡的能屏障。因此,氢原子保持在其初始位置的附近,从而导致模拟时间尺度内未平衡的成对分布。这些结果表明,尽管该系统在较高温度下接近随机分布,但要在室温下实现平衡和泊松行为,以实现较长的模拟,以实现低弹性状态。
讨论
总而言之,我们的研究使我们能够通过严格有效的方法在各种温度下在结构化环境(例如纯镁)内彻底表征氢的动力和迁移率。我们在不同的训练模式下(通用和微调),对基于ML的原子间潜在MD方案进行了系统和比较研究,尤其是针对贝叶斯对贝叶斯与均衡神经网络(MACE和CHGNET)(MACE和CHGNET)(MACE和CHGNET)(MACE和CHGNET)。通过对扩散系数和激活能值的估计来验证结果,这表明与实验数据一致。获得的结果证明,在MLFF训练期间收集的VASP-DFT构型代表了氢和镁原子之间原子质相互作用的准确建模的完整集。可以始终应用此策略来生成一个全面的数据集,以适当培训现有或即将发行的潜力。实际上,由于其数据集中缺乏代表性的高温,有缺陷且可亚稳态的状态,我们确定了预训练的UIP在研究具有扩散缺陷的材料中的局限性。特别是,在对积极学习的DFT配置进行了微调之后,我们展示了最先进的机器学习模型实现DFT级准确性的能力,突出了将重点放在有效的数据集构建方法及其质量上的重要性。具体而言,包括缺陷和过渡状态配置以及转移学习的作用的重要性,从而允许预先训练的解决方案适应新系统。所获得的数据为不同的氢浓度时提供了对集体动力学特性的宝贵新见解,可以准确预测随着浓度的增加,氢迁移率会降低。此外,氢对分布的统计分析表明该系统在较高温度下接近随机分布。但是,在室温下,在低驾驶状态下,在室温下实现平衡和泊松行为是必要的。
这些进展不仅可以提高我们对镁氢相互作用的理解,而且还为将来的研究铺平了多种多样的多组分系统和缺陷组成的道路。这对于具有复杂势能表面的系统尤其重要,其中传统的ABInitio方法变得不切实际。通过机器学习加速分子动力学成功地对镁中的氢扩散机制进行建模,可以促进并发现新的,更有效的材料,以供氢存储及其储存,从而有助于向更绿色,更可持续的能源未来的过渡。
方法
MLFF-MD
密度功能理论(DFT),MLFF-MD和攀爬图像-NEB39(Cineb)使用VASP版本6.4.2和版本6.4.3进行计算33,,,,34,,,,40,,,,41, 分别。我们利用了4ââ€4超级电池,如图所示。5,在HCP晶体对称性中包含128 mg原子,具有8小时原子(MGH)0.0625)随机分布在晶格中。我们在Perdew-Burke-Ernzerhof(PBE)的理论水平上进行了非自旋偏振计算42,使用600 eV的能量截止,4 4 k点网格,收敛阈值为0.1 meV,使用的高斯涂片为0.05 ev。为了确保一致性并避免与数据库的任何差异,我们采用了与材料项目数据库中使用的收敛参数相同的收敛参数。对于VASP-MLFF,我们使用了默认参数。在0.2 ns间隔内使用VASP在正式的MLFF-MD上,温度从0升至700 k。在此设置中,用于训练原子间电位的设置,包括作用在每个原子,原子坐标,应力张量和晶格参数上的力的力。每当贝叶斯错误超过集合固定阈值时,VASP都会恢复为DFT以在数据库中生成新的配置。这种即时的过程允许模型使用累积的AB-Initio配置来逐渐改善后续步骤的预测。已设置了5âmeV/ã的阈值值,以确保从探索的配置相位空间中收集数据库中结构的各种且广泛的表示。在热化阶段,我们在限制细胞形状的同时选择了NPT集合1。我们施加了零外部压力,时间步长为1 fs。在300 K,480 K和673 K的关键温度下,我们在训练模式下进行了100 ps长的NPT模拟,以积累其他配置。总共存储了超过3700个AB-Initio配置,在每个恒定温度下记录的少于1000,并且在坡道阶段捕获的其余配置。随后,我们在运行模式下切换到MLFF-MD,并确定了300 k,480 k和670 k的固定温度下100 ps期间的平均晶格体积。然后将平均体积配置用于进行NVT模拟。在100 ps NVT模拟之后,我们提取了三个结构,其能量接近NVT运行的平均值。为了避免任何相关性,我们确保它们之间的时间间隔至少为10 ps。随后,从这些开始,我们进行了三个不同的NVE模拟,在其中计算了氢原子的平均平方位移(MSD)作为集合平均值
$$ {\ rm {msd}}(t)= \ frac {1} {t-t} \ mathop {\ int} \ nolimits_ {0}^{t-t-t-t} {[{\ bf {\ bf {r}}(r}}(t+\ delta) -)]}^{2} d \ delta。$$
(2)
在哪里t是总模拟时间,并且r是正在分析的原子的轨迹。在最后的NVE运行期间,采用了0.5 fs的时间步长以减少能量波动。所有MSD均在1 ns的NVE轨迹上构建,并用于提取扩散系数d通过将功能的线性部分与爱因斯坦关系拟合$$ {\ rm {msd}}}(t)= 6dt $$
(3)
通过拟合模拟的初始0.2 NS线性区域。
价值d在每个温度下,计算为三个NVE复制品的平均值。相关的不确定性计算为平均SEM = SD/的标准误差\(\ sqrt {3} \),其中SD是标准偏差,3我们收集的预测的数量。MSD拟合程序的一个示例包括在补充图中。SF1。

Mg原子以橙色显示为粉红色,H原子显示。
Cineb计算是使用2ââ€2â€2超级细胞进行的,其中一个氢位于文献中鉴定出的间隙位点之间的主要路径17。在补充图中报道了这些位点的可视化。SF3。同样,每次计算都使用十个图像和0.01 ev/veral收敛标准进行。
通用的原子间电位UIP
我们考虑两个最先进的表现43预训练的UIP:狼牙棒44,基于一条通过神经网络和CHGNET31,基于图的神经网络。这些模型是在材料项目(MP)放松轨迹数据库中进口的预培训版本45,包括160万个具有相关能量,力,应力和磁矩的晶体结构。为了解决此类UIP的预测动力学特性,我们遵循了与上一节所述的相同的NEB和MD程序(不包括在线训练阶段)。随后,在fly MLFF-MD期间获得的VASP-DFT生成的数据对CHGNET和MACE进行了微调。梅斯还在数据集中从头开始训练。所有模型的训练和微调都是使用相应GitHub存储库中开发人员建议的标准程序和参数进行的。在CHGNET的MD模拟过程中,我们观察到需要0.5 fs的时间步长以稳定NPT和NVT运行中的动力学,从而避免了NVE温度的漂移。在这方面,我们显示了在补充无花果中的0.5 fs和1.0 fs病例的NVE模拟过程中温度振荡的示例。SF4和SF5。为了执行MD模拟,我们采用了lammps46和ASE47,分别用于梅斯和Chgnet。所采用的工作流程的示意图如图所示。6。此外,使用最佳性能模型(MACE_FT),我们遵循相同的协议进行估算d在三个额外的氢浓度下,含10小时(MGH)0.078125),6 h(MGH0.046875)和4 h(MGH0.03125), 分别。为了评估每个模型的性能,我们评估了均方根误差(RMSE),以预测能量和力在1200个配置的测试数据集上。从MACE_FT模型的NVE复制品中随机对它们进行随机采样,并在每个温度中使用400个样品。This validation process was independently repeated for each different hydrogen content to test the models’ accuracy beyond the training concentration.At last, NEB calculation on the same path studied using VASP were performed with every model using the ciNEB implementation of ASE employing the same number of images and convergence criteria.

The database of configurations is built both during NpT-MD thermalizations of the system from 0 K to 700 K via active learning of the VASP-MLFF, and at target temperatures (300 K, 480 K, and 673 K).Subsequently, the machine learned potentials are fine-tuned or trained, and after system-equilibration the MSD and the diffusion coefficient D at fixed T are computed.
参考
Yang, J., Sudik, A., Wolverton, C. & Siegel, D. J. High capacity hydrogen storage materials: attributes for automotive applications and techniques for materials discovery.化学Soc。修订版 39, 656–675 (2010).
CAS一个 PubMed一个 Google Scholar一个
Bhimineni, S. H. et al.Machine-learning-assisted investigation of the diffusion of hydrogen in brine by performing molecular dynamics simulation.Ind。Eng。化学res。 62, 21385–21396 (2023).
CAS一个 Google Scholar一个
Tarasov, B. P. et al.Metal hydride hydrogen storage and compression systems for energy storage technologies.int。J. Hydrog.活力 46, 13647–13657 (2021).
CAS一个 Google Scholar一个
Turner, J. A. A realizable renewable energy future.科学 285, 687 - 689 (1999).
PubMed一个 Google Scholar一个
Gardner, D. Hydrogen production from renewables.可再生能源重点 9, 34–37 (2009).
Aceves, S. M., Berry, G. D., Martinez-Frias, J. & Espinosa-Loza, F. Vehicular storage of hydrogen in insulated pressure vessels.int。J. Hydrog.活力 31, 2274–2283 (2006).
CAS一个 Google Scholar一个
Sun,Y。等。Tailoring magnesium based materials for hydrogen storage through synthesis: Current state of the art.储能母校。 10, 168–198 (2018).
Felderhoff, M., Weidenthaler, C., von Helmolt, R. & Eberle, U. Hydrogen storage: the remaining scientific and technological challenges.物理。化学化学物理。 9, 2643–2653 (2007).
CAS一个 PubMed一个 Google Scholar一个
Hua, T. et al.Technical assessment of compressed hydrogen storage tank systems for automotive applications.int。J. Hydrog.活力 36, 3037–3049 (2011).
CAS一个 Google Scholar一个
美国能源部。DOE technical targets for onboard hydrogen storage for light-duty vehicles.https://www.energy.gov/eere/fuelcells/doe-technical-targets-onboard-hydrogen-storage-light-duty-vehicles(2023)。
Yartys, V. et al.Magnesium based materials for hydrogen based energy storage: past, present and future.int。J. Hydrog.活力 44, 7809–7859 (2019).
CAS一个 Google Scholar一个
Hirscher, M. et al.Materials for hydrogen-based energy storage - past, recent progress and future outlook.J.合金。compd。 827, 153548 (2020).
CAS一个 Google Scholar一个
KlopÄiÄ, N., Grimmer, I., Winkler, F., Sartory, M. & Trattner, A. A review on metal hydride materials for hydrogen storage.J.储能 72, 108456 (2023).
Allendorf, M. D. et al.Challenges to developing materials for the transport and storage of hydrogen.纳特。化学 14, 1214–1223 (2022).
CAS一个 PubMed一个 Google Scholar一个
Gupta, A. & Faisal, M. Magnesium based multi-metallic hybrids with soot for hydrogen storage.int。J. Hydrog.活力 53, 93–104 (2024).
CAS一个 Google Scholar一个
Schimmel, H., Kearley, G., Huot, J. & Mulder, F. Hydrogen diffusion in magnesium metal (αphase) studied by ab initio computer simulations.J.合金。compd。 404-406, 235–237 (2005).
CAS一个 Google Scholar一个
Klyukin, K., Shelyapina, M. G. & Fruchart, D. Dft calculations of hydrogen diffusion and phase transformations in magnesium.J.合金。compd。 644, 371–377 (2015).
CAS一个 Google Scholar一个
Shelyapina, M. G. Hydrogen diffusion on, into and in magnesium probed by dft: a review.氢3 , 285–302 (2022).CAS
一个 Google Scholar一个 Nishimura, C., Komaki, M. & Amano, M. Hydrogen permeation through magnesium.J.合金。
compd。293-295 , 329–333 (1999).CAS
一个 Google Scholar一个 Uchida, H. et al.Absorption kinetics and hydride formation in magnesium films: effect of driving force revisited.
Acta Mater。85 , 279–289 (2015).CAS
一个 Google Scholar一个 Renner, J. & Grabke, H. J. Bestimmung von diffusionskoeffizienten bei der hydrierung von legierungen.int。
J. Mater。res。 69, 639–642 (1978).
CAS一个 Google Scholar一个
Kwon, H., Shiga, M., Kimizuka, H. & Oda, T. Accurate description of hydrogen diffusivity in bcc metals using machine-learning moment tensor potentials and path-integral methods.Acta Mater。 247, 118739 (2023).
CAS一个 Google Scholar一个
Tang,H。等。Reinforcement learning-guided long-timescale simulation of hydrogen transport in metals.ADV。科学。 11, 2304122 (2024).
CAS一个 Google Scholar一个
Kimizuka, H., Thomsen, B. & Shiga, M. Artificial neural network-based path integral simulations of hydrogen isotope diffusion in palladium.J. Phys。活力 4, 034004 (2022).
CAS一个 Google Scholar一个
Lu, G. M., Witman, M., Agarwal, S., Stavila, V. & Trinkle, D. R. Explainable machine learning for hydrogen diffusion in metals and random binary alloys.物理。牧师。 7, 105402 (2023).
CAS一个 Google Scholar一个
Friederich, P., Häse, F., Proppe, J. & Aspuru-Guzik, A. Machine-learned potentials for next-generation matter simulations.纳特。母校。 20, 750–761 (2021).
CAS一个 PubMed一个 Google Scholar一个
Yu, F., Xiang, X., Zu, X. & Hu, S. Hydrogen diffusion in zirconium hydrides from on-the-fly machine learning molecular dynamics.int。J. Hydrog.活力 56, 1057–1066 (2024).
CAS一个 Google Scholar一个
Vandenhaute, S., Cools-Ceuppens, M., DeKeyser, S., Verstraelen, T. & Van Speybroeck, V. Machine learning potentials for metal-organic frameworks using an incremental learning approach.npj Comput.母校。 9, 1–8 (2023).
Kývala, L., Angeletti, A., Franchini, C. & Dellago, C. Diffusion and coalescence of phosphorene monovacancies studied using high-dimensional neural network potentials.J. Phys。化学c 127, 23743–23751 (2023).
Wang, N. & Huang, S. Molecular dynamics study on magnesium hydride nanoclusters with machine-learning interatomic potential.物理。修订版b 102, 094111 (2020).
CAS一个 Google Scholar一个
Deng, B., Zhong, P. & Jun, K. et al.Chgnet as a pretrained universal neural network potential for charge-informed atomistic modelling.纳特。马赫。Intell。 5, 1031–1041 (2023).
Batatia, I., Kovacs, D. P., Simm, G., Ortner, C. & Csanyi, G. Mace: higher order equivariant message passing neural networks for fast and accurate force fields.在神经信息处理系统的进步,卷。35 11423–11436 (eds Koyejo, S. et al.) (Curran Associates, Inc., 2022).
Jinnouchi, R., Karsai, F., Verdi, C., Asahi, R. & Kresse, G. Descriptors representing two- and three-body atomic distributions and their effects on the accuracy of machine-learned inter-atomic potentials.J. Chem。物理。 152, 234102 (2020).
CAS一个 PubMed一个 Google Scholar一个
Jinnouchi, R., Karsai, F. & Kresse, G. On-the-fly machine learning force field generation: application to melting points.物理。修订版b 100, 014105 (2019).
CAS一个 Google Scholar一个
Verdi, C., Ranalli, L., Franchini, C. & Kresse, G. Quantum paraelectricity and structural phase transitions in strontium titanate beyond density functional theory.物理。牧师。 7, L030801 (2023).
CAS一个 Google Scholar一个
Ranalli, L. et al.Temperature-dependent anharmonic phonons in quantum paraelectric ktao3 by first principles and machine-learned force fields.ADV。量子技术。 6, 2200131 (2023).
CAS一个 Google Scholar一个
Verdi, C., Karsai, F., Liu, P., Jinnouchi, R. & Kresse, G. Thermal transport and phase transitions of zirconia by on-the-fly machine-learned interatomic potentials.npj Comput.母校。 7, 156 (2021).
CAS一个 Google Scholar一个
Liu, P., Verdi, C., Karsai, F. & Kresse, G. Phase transitions of zirconia: machine-learned force fields beyond density functional theory.物理。修订版b 105, L060102 (2022).
CAS一个 Google Scholar一个
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths.J. Chem。物理。 113, 9901–9904 (2000).
CAS一个 Google Scholar一个
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set.物理。修订版b 54, 11169–11186 (1996).
CAS一个 Google Scholar一个
Kresse,G。&Joubert,D。从Ultrasoft伪电势到投影仪增强波的方法。物理。修订版b 59, 1758–1775 (1999).
CAS一个 Google Scholar一个
Perdew, J. P., Burke, K. & Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system.物理。修订版b 54, 16533–16539 (1996).
CAS一个 Google Scholar一个
Riebesell, J. et al.Matbench discovery–an evaluation framework for machine learning crystal stability prediction.Preprint athttps://arxiv.org/abs/2308.14920(2023)。
Batatia, I. et al.A foundation model for atomistic materials chemistry.Preprint athttps://arxiv.org/abs/2401.00096(2023)。
Jain,A。等。Commentary: The materials project: a materials genome approach to accelerating materials innovation.apl mater。 1, 011002 (2013).
Thompson, A. P. et al.LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales.计算。物理。社区。 271, 108171 (2022).
CAS一个 Google Scholar一个
Larsen, A. H. et al.The atomic simulation environment-a python library for working with atoms.J. Phys。冷凝物。事情 29, 273002 (2017).
致谢
C.F.and A.A.acknowledge the “Doctoral College Advanced Functional Materials - Hierarchical Design of Hybrid Systems DOC 85 doc.funds†funded by the Austrian Science Fund (FWF) and by the Vienna Doctoral School in Physics (VDSP).For Open Access purposes, the author has applied a CC BY public copyright license to any author accepted manuscript version arising from this submission.D.M.and S.P. were supported by the European Union Horizon 2020 research and innovation program under Grant Agreement No. 857470, from the European Regional Development Fund under the program of the Foundation for Polish Science International Research Agenda PLUS, grant No. MAB PLUS/2018/8, and the initiative of the Ministry of Science and Higher Education ’Support for the activities of Centers of Excellence established in Poland under the Horizon 2020 program’ under agreement No. MEiN/2023/DIR/3795.L.P. and C.F.acknowledge the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3 - Project NEST (Network 4 Energy Sustainable Transition) of Ministero dell’Universitá e della Ricerca (MUR), funded by the European Union - NextGenerationEU.L.L. and C.F.acknowledge the NRRP, CN-HPC grant no.(CUP) J33C22001170001, SPOKE 7, of MUR, funded by the European Union - NextGenerationEU.The computational results were obtained using the Vienna Scientific Cluster (VSC) and the LEONARDO cluster.We acknowledge access to LEONARDO at CINECA, Italy, via an AURELEO (Austrian Users at LEONARDO supercomputer) project.
道德声明
竞争利益
作者没有宣称没有竞争利益。
附加信息
出版商的注释关于已发表的地图和机构隶属关系中的管辖权主张,Springer自然仍然是中立的。
补充信息
权利和权限
开放访问This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.本文中的图像或其他第三方材料包含在文章的创意共享许可证中,除非在材料的信用额度中另有说明。如果文章的创意共享许可中未包含材料,并且您的预期用途不得由法定法规允许或超过允许的用途,则需要直接从版权所有者那里获得许可。要查看此许可证的副本,请访问http://creativecommons.org/licenses/4.0/。重印和权限
引用本文

Angeletti, A., Leoni, L., Massa, D.
等。Hydrogen diffusion in magnesium using machine learning potentials: a comparative study.npj Comput Mater11 , 85 (2025).https://doi.org/10.1038/s41524-025-01555-z
已收到:
公认:
出版:
doi:https://doi.org/10.1038/s41524-025-01555-z