
集成机器学习驱动的虚拟筛选和分子动力学模拟,以鉴定针对PARP1针对前列腺癌的潜在抑制剂
作者:Dabwan, Khaled H.
介绍
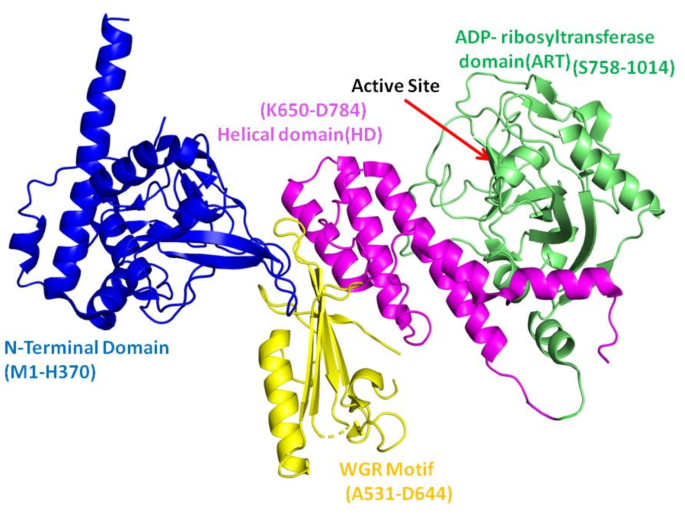
前列腺癌(PC)是男性最普遍的恶性肿瘤形式之一,近年来新诊断的病例显着增加1。每年,这种疾病会杀死35万多人2,,,,3。聚(ADP-核糖)聚合酶1(PARP1)是一种无处不在的核酶,在多种核功能中发挥作用,包括DNA修复,核糖体合成,核糖素调节,核转运,核转运和表观遗传学书签(图) 1)。激活时,PARP1捕获了NAD+,并组装了聚(ADP-核糖)的长聚合物,共价改变自身和周围的核蛋白4,,,,5,,,,6。PARP1最普遍,最有良好的动作之一是DNA修复。当与BRCA1,BRCA2,PTEN,PTEN,ATM,CHEK2和FANCA的几个DNA修复基因的活性丧失时,PARP1抑制可能对突变细胞致命7,,,,8,,,,9。因此,PARP1抑制剂可以特异性地靶向DNA修复机制异常异常的肿瘤细胞10,,,,11。PARP1具有特定的域设计,在DNA修复和其他细胞功能中起着重要作用。PARP1由三个结构域组成,例如C末端催化结构域(CD),DNA结合结构域(N末端)和中心结构域。N末端结构域由识别DNA损伤并有助于与DNA结合的三个锌指基序组成。PARP1的中心结构域含有充当ADP-核糖部分受体的谷氨酸和赖氨酸残基,以及允许与其他DNA损伤响应蛋白相互作用的BRCT结构域12。催化结构域(CAT)由两个子域组成:螺旋结构域(HD)和ADP-核糖基转移酶(ART)结构域,在ART域中具有活性位点。这些域的折叠架构在所有PARP家庭成员中都保存13,,,,14,,,,15。该催化结构域对于靶蛋白的聚(adpribosyl)(adpribosyl)ation(Parylation)至关重要,该靶蛋白使用NAD+ - 形成聚(ADP-核糖)链以进行DNA修复和细胞调节。

PAPR1(PDB ID 4DQY)的域除外。在这种晶体结构中不存在BRCT结构域。催化结构域的子域被涂成WGR(黄色),螺旋域(粉红色)和Art域(绿色)。使用Pymol 3.0.4绘制图像(https://www.pymol.org/),标签是在Microsoft Office Power Point中进行的。
实验研究表明,某些残基,例如D766,E763,D770,Y907,R878,S864,H862,K903和SER904,对于PARP催化作用至关重要。这些残基对酶活性至关重要,因为它们直接参与催化过程和酶底物复合物的稳定性16。Talazoparib和Olaparib是PARP1抑制剂的实例,它们有选择地靶向PARP1酶的催化区域。这些抑制剂会导致癌细胞,尤其是患有BRCA突变的癌细胞,会在癌细胞中积累损伤DNA。除此之外,还有多个研究研究了PARP1对PC的效果抑制剂17,,,,18,,,,19。由于同源重组是BRCA突变受损的DNA修复中的关键步骤,因此细胞严重依赖于PARP1来修复DNA损伤20。通过PARP1抑制,这些药物可以阻止DNA损伤被修复,从而杀死癌细胞。这些药物通过阻止PARP1产生合成的杀伤力,导致细胞死亡,并显示出对BRCA突变的恶性肿瘤的重要潜力21根据当前的研究和临床研究,针对PARP1的ADP-核糖基转移酶结构域(ART)的抑制剂有助于治疗具有DNA损伤治疗基因突变的PC癌症11,,,,22。在PC中,雄激素受体(AR)转录活性需要PARP1。在被AR靶位点吸引后,PARP1酶活性在AR驱动的基因表达和PC细胞的生长中起着至关重要的作用。抑制PARP1可减少AR转录活性,从而影响雄激素依赖性和非依赖性AR激活。PARP1在前列腺癌中的显着性,尤其是在AR信号传导和DNA修复途径中,其作为治疗靶标的潜力11。计算生物学彻底改变了药物发现和开发,机器学习驱动的虚拟筛选和分子动力学模拟作为强大的工具出现。机器学习算法可以有效地处理和分析庞大的化学文库,从而可以高精度预测潜在的抑制剂。这些算法使用已知抑制剂的模式和特征来识别有效结合靶蛋白的新化合物23,,,,24。将机器学习驱动的虚拟筛选与分子动力学模拟整合在一起,为识别靶向PARP1的ADP-核糖基转移酶结构域(ART)的潜在抑制剂提供了一种有希望的策略,在前列腺癌中,有可能改善治疗结果并提供新的途径,以抗击这种普遍和致命的疾病。机器学习(ML)是药物发现的强大工具,可以基于功效和类似药物的特性快速筛选化合物的快速筛查和优先次序。当与分子动力学模拟结合使用时,它显着提高了抑制剂鉴定的精度和效率25,,,,26。在这项研究中,与分子动力学模拟集成的基于基于的虚拟筛选和分子对接被用于鉴定潜在的PARP1抑制剂。筛选了一个植物化学物质的文库,重点关注那些对PARP1的药物能力高和潜在抑制作用的库。
方法
收集和处理数据
从BINDINGDB中检索到靶向PARP1酶的6,510个活性抑制剂的综合文库27。有用的诱饵增强目录(dud-e)(dud-e)(https://dude.docking.org/(在2024年9月16日访问)资源用于创建一组2871个诱饵化合物28。为了维持数据完整性,仔细发现并消除了活性抑制剂数据集中的重复化合物。为了使更多的实验研究更容易,然后将诱饵和主动抑制剂组合成单个CSV文件。从BindingDB下载的活动抑制剂的值为1,表示其活动,并标记为0值,表示其不活动,以创建基于机器学习的预测模型。在模型训练过程中,这种标记方案对于区分活性分子和非活性分子至关重要。
分子描述符的生成
使用RDKIT计算数据集的2D分子描述符(https://www.rdkit.org/),一个流行的开源化学信息学计划。最初开发了总共34个描述符,其中包括各种化学特征和结构方面。为了验证数据集的完整性和质量,严格消除了无效值的描述符。在此预处理阶段之后,进行了特征选择,以保留最相关和最有用的描述符。此方法保证数据集具有用于模型培训和测试的高质量功能。图 2描述了研究方法的步骤,说明了从数据集准备到基于ML的筛选和分子对接的每个步骤。

研究的工作流程。这个数字是使用Microsoft Office Power Point创建的。
主成分分析(PCA)
为了处理数据的较大维度,使用了主成分分析(PCA)。PCA是一种标准且经常使用的用于模式检测和数据表示的方法。它将原始的高维数据集转换为较低维空间,同时保留大多数重要信息。维度下降是通过识别主要组件来实现的29。为了执行PCA,生成数据集的协方差矩阵以确定最高方差的方向。计算特征值和相关特征向量,以量化和表征每个主要成分的重要性。具有较高特征值的组件对数据集的差异做出了巨大贡献,并优先考虑进一步研究。
机器学习模型
开发和测试了许多机器学习模型,以确定数据集中的化合物是活性还是不活动。这些模型是在处理后的数据集上创建的。这项研究中采用的算法是支持向量机(SVM),K-Nearest邻居(KNN),幼稚的贝叶斯(NB)和随机森林。SVM,K-Nearest邻居,Na-Ve贝叶斯和随机森林在药物发现中是流行的算法,因为它们在高维空间中的有效性,对复杂数据集的分类,简单性和分子活性的预测。SVM在用于抑制剂识别的虚拟筛查研究中特别有用,KNN在QSAR建模中简单有效,随机森林是稳健,准确且可抵抗过度拟合的30,,,,31,,,,32。SVM方法以其在高维环境中的效率和对过度拟合的抵抗而闻名,尤其是在尺寸比样品更大的尺寸时。SVM确定以最好的方式将数据分为两类的超级平面,从而给出了两类之间最大的余量。KNN是一种直接但有效的方法,它使用了描述符空间中其最近的邻居的多数类,以确定化学物质是活性还是无活性。这是一种非常简单的方法,它基于以下想法:类似化合物可能具有相似的属性。基于贝叶斯定理,NB概率分类器假定每个特征都是独立的。在许多实际应用中,NB的运作效果非常简单,有时是不现实的特征独立性假设。这使其成为化学信息学方面的宝贵工具。RF是一种合奏学习方法,它在训练过程中构建许多决策树,并输出代表班级模式或单个树的平均预测的类。随机森林以其出色的准确性,能够以增加维度来处理大数据集的能力以及对过度安装的弹性而闻名。
模型评估
在这项工作中,我们使用十倍的交叉验证来彻底测试机器学习模型的性能。这种可靠的验证方法将数据集划分为10个独特的子集或折叠。使用多种统计措施评估模型功效,包括准确性,灵敏度,特异性和MCC。模型的准确性被计算为在整个实例中正确预测实例(主动和非活动)的比例。
$$准确= \ frac {tp + tn} {{tp + tn + fp + fn}} $$
(1)
特异性被测量为
$$特异性= \ frac {tn} {{{tn + fp}} $$
(2)
灵敏度估计为:
$$敏感性{} = \ frac {tp} {{tp + fn}} $$
(3)
这些绩效指标经过严格计算,以对每个模型的预测能力进行彻底评估。包含许多统计因素提供了有关模型优势和限制的完整知识,最终指导了选择最成功的预测模型,以区分活性和非活动化合物。
基于机器学习模型筛查
由9510植物化学物质组成的库来自锌,Pubchem和Chembl数据库,以进行虚拟筛选。先前针对我们初始数据集建立的处理和功能计算方法系统地应用于此新数据集。然后使用我们的预训练的机器学习模型对处理后的库进行筛选,该模型将每种化合物识别为活动性或无活性。此外,使用Lipinski的五个规则评估了筛选的活跃化合物。
分子对接
基于机器学习模型筛选后,将筛选的化合物停靠到PARP1的活动位置。用Autodock Vina进行了分子对接实验。Autodock Vina是广泛使用的对接软件,可预测化合物与受体的结合。它通过分析配置并根据绑定亲和力对它们进行评分,从而有效地寻找最佳的结合姿势33。从PDB ID 5DS3下的PDB数据库中检索了猫域的ART子域的晶体学结构(https://www.rcsb.org/structure/5ds3)。使用AutoDockTools-1.5.6制备蛋白质和化合物。极性氢键和充电的蛋白质和化合物计算。后来,在化合物上,蛋白质以PDBQT格式保存,用于对接实验。对接实验的网格大小设置为x,y和Z的45ââ,而盒子则以Center_x = –16.759,Center_y = 30.743和Center_z = Center_z = center_ = - 132.285。选择这种尺寸是为了覆盖完整的活跃部位,同时为配体提供了探索相邻绑定区域的自由。每种化合物的详尽参数设置为16和20数的姿势。
分子动力学(MD)模拟
在这项研究中,我们使用了gromacs 2021.334为了模拟apo形式和源自自动库克vina的复合物。CHARMM36-JUL202234用于蛋白质拓扑,而使用CGENFF服务器生成配体拓扑35。将系统用TIP3P水模型溶解在一个立方盒中,以确保从盒子的每一侧将蛋白质定位为15â。引入了钠离子(Naâ+)以中和系统。中和后,使用最陡峭的下降方法执行了1000步的最小化。然后,使用NVT和NPT集合以300 ps稳定温度和压力,对系统进行平衡。每个系统都进行了100 ns模拟,并保存了轨迹以进行后续分析。LINCS算法用于对键长的约束,并将约束应用于涉及氢原子的键。每10 ps后保存轨迹。对根平方偏差(RMSD),均方根波动(RMSF)和循环半径(RG)进行了详细分析。
自由能分析(MM-PBSA)
模拟完成后,计算PARP1和化合物复合物的结合能,以评估复合物的稳定性。这项研究对于理解生物分子相互作用及其成分至关重要。GMX_MMPBSA模块用于进行这些计算。对于MM-PBSA计算,提取了100 ns模拟的最后25 ns的5000帧。该应用通过分析多种结合成分(例如极性,非极性,结合和非键相互作用)来计算蛋白配体复合物的结合能。
使用以下公式估算复合物的结合自由能:
$$ \ delta g_ {binding} = \ delta g_ {complect} - \ left({\ delta g_ {protein} + \ delta g_ {ligand}}} \ right)$$
(4)
在这个等式中\(\ delta g_ {binding} \)代表无网络结合能\(\ delta g_ {复杂} \)是配体蛋白质复合物的结合自由能,\(\ delta g_ {蛋白质} \)是蛋白质的自由能,\(\ delta g_ {ligand} \)是溶剂中配体的自由能。自由能也可以表示为:
$$ \ delta g_ {binding} = \ delta e_ {mm} + \ delta e_ {solv} -t \ delta s $$
(5)
在等式中(5)\(\ delta e_ {solv} \)是由极性溶剂化(EGB)和非极性溶剂化能(ESURF)组成的溶剂化自由能。\(\ delta e_ {mm} \)包括非键的能量静电elec和van der waals相互作用 - EVDW和键合的能量也称为内部能量,包括角度弯曲,键拉伸和扭转相互作用。\(t \ delta s \)表示熵能贡献。MM-PBSA方法通过省略熵项来计算有效的结合能。当排除熵项时,估计值是有效的自由能,通常足以比较相关配体的结合自由能36,,,,37。GMX_MMPBSA模块系统地评估了这些组件,从而对绑定相互作用提供了全面的描述。
毒性分析
化学毒性评估是药物开发过程中的重要步骤。除了更快外,毒性估计的计算技术还可以减少动物测试的需求。研究人员可能会通过发现开发过程早期毒性问题较少的化合物来降低药物研究和开发中经常观察到的高损耗率。Protox-II38WebServer用于评估所选化合物的毒性。Protox-II服务器提供了一个完整的平台,用于预测终点毒性的多样性,包括诱变,细胞毒性,肝毒性和致癌性。
结果
机器学习模型生成的数据收集和预处理
分别从BindingDB和DUD-E检索了6510个活性的数据集和2871个针对PARP1的诱饵化合物。然后将活性化合物数据进行预处理,以去除重复的条目,总共产生4046种化合物。数据集中的活性化合物被标记为1,并标记为0。集成数据集由6917种化合物(4046个活性剂和2871个无效物)组成,分为训练和测试集,分别为4841(70%)(70%)和2076(30%)复合(分别)(分别)。1)。表1火车和测试集的统计数据。
最初,使用RDKIT确定了从上一步中选择的化合物的可量化的2D分子和结构描述。
总共计算了34个描述符。对于模型训练,不包括零值的描述符。表中列出了针对数据集计算的符号的列表S1。针对药物发现的描述符的选择是基于它们与吸毒,生物学活性和物理化学特性的相关性。主要描述符,例如分子量,LOGP,氢键供体,受体,TPSA和可旋转键,因为它们对吸收,分布,代谢,排泄和毒性曲线的影响。这些属性与诸如Lipinski的五个规则等准则保持一致39和Veber的规则40。
原理分析
我们使用PCA降低了数据集的维度,该数据集包含34个描述符的不同物质。通过使用PCA,我们能够将数据凝结到两个主要组件中,每个组件都将原始描述符组合在一起,以代表化学物质的最重要的基础模式和特征。图 3说明了PCA的前两个主要成分的散点图。在PCA散点图中,标签1(活性化合物)和标签0(非活性化合物)的重叠区域表示化合物具有相似描述符值的区域。这表明这些区域中的活性和非活性化合物具有可比的化学或结构特征,使它们更具挑战性,仅基于所选描述源来区分。第一个组件的特征值为1863.27,第二个组件为5.28。这些值代表了每个组件在数据集中所占的差异数量。特征值越高,组件提取的差异越多。在我们的示例中,PC1具有较高的特征值,占差异的99.59%,捕获了我们最初的高维数据中隐藏的最高关键信息。PC2贡献了额外的0.28%的解释变化。这些差异表明这两个主要成分有效地保留了数据集的重要信息。PCA使得可以通过最小化尺寸的数量来执行更简单的分析和可视化,而不会损害数据集中的关键信息。

2个主要组成部分的情节。红色和蓝色表示活性和非活性化合物。
化学空间和多样性分析
机器学习模型的性能受培训和测试数据集中存在的化学异质性的影响。从不同的样本种群看来,开发的模型可能在新的数据上表现出色。在当前的研究中,样品的生理化学分布以分子量和logP表示,并绘制在图的两个轴上(图。 4)。选择分子量和LOGP进行化学空间研究,因为它们是类似药物的特性的重要描述和Lipinski五个规则的关键组成部分。这些参数可直接评估数据集的多样性,同时着重于分子大小和亲脂性,这对于吸收,分布和靶点结合都很重要。分子量分别在70至1562 Daltons和70至1313 Dalton之间进行测试和训练集。对于测试和训练集,LOGP的值分别在3.5至9.0和5.20至10之间变化。

训练集和测试集中多样性的分布,由诸如LOGP和分子量等参数定义。
生成模型和验证
为了构建模型,应用了四种监督学习算法(KNN,NB,RF和SVM)。这些模型是使用Python Scikit-Learn库开发的,并计算了各种统计措施(准确性,灵敏度,特异性,MCC和曲线下的区域),以评估每个模型的性能。这些评估指标的值显示了2。根据性能索引,很明显,RF胜过所有具有最高准确性值(0.948940),特异性(0.917056),MCC(0.894556)和AUC(0.984607)的模型。在KNN和SVM之间观察到A TIE,具有较高的精度值(0.906551 vs 0.905106),特异性(0.809579 vs 0.806075)和MCC(0.809970 vs 0.807131)。但是,与KNN相比,SVM的AUC(0.963942)更高(0.943203)。Na -Ve Bayes表现出最差的性能,其值最低,该值对所有计算的参数(准确性0.827553,灵敏度0.877049,MCC 0.641622和AUC 0.874420)表现出最差的表现。与上述发现一致,RF分别在训练和测试数据集中分别形成了ROC-AUC曲线的最高峰,然后是KNN和SVM(图。 5)。灵敏度是模型正确识别阳性的能力,该阳性描绘了偏爱KNN和SVM(0.974590)的趋势(0.971311)。结论性地,RF模型被认为是最好的,并被用于筛选9000个植物化学物质的文库,以预测活性分子。机器学习算法的选择是基于它们在处理化学信息学数据方面的公认有效性。RF因其处理高维数据集的能力,防止拟合的鲁棒性以及捕获非线性关系的能力而被选择。RF算法已被广泛应用于药物发现中,以预测分子活性并确定潜在的抑制剂41,,,,42。

测试和火车集中所有模型的ROC-AUC曲线显示了每个模型的性能。
药物相似性
在9000个中,发现了181种对PARP1的活性。随后,仅采用五个规则来入围具有良好渗透和吸收潜力的化合物。选择了分子量> 500DALTON的化合物,<5氢键供体,<10个氢键受体和5个氢键受体和5个<logP <forp -<5,以进行进一步的对接研究和治疗性评估作为有效的药物候选者。在181种化合物中,有40种符合所有四种毒品相似标准。绘制了热图以研究活性化合物的分子特性之间的相关性(图。S2)。在氢键受体(HBA) - 氢键供体(HBD)[0.229]和LOGP-HBD [0.291]之间见证了浅蓝色指示的两个正相关。
分子对接
使用Olaparib作为参考,评估了筛选的药物(如化合物)与PARP1活性位点的结合相互作用。使用Autodock Vina,将所有40个分子(包括参考)都停靠在PARP1活动位置。每个对接实验都获得了十个对接姿势。基于能量评分排列化合物。所有化合物的结合能介于10.8 kcal/mol和6.3 kcal/mol之间。然而,锌43120769(10.8 kcal/mol),锌40934164(10.2 kcal/mol)和Zinc14584870(9.9 kcal/mol)(表)(表)(表)(表)(表)3)。这些化合物在PARP1的装订袋中与参考的同一位置。参考化合物Olaparib与Ser904 Gly863,arg878和Tyr896表现出氢键相互作用(图。S1)。复杂的锌43120769建立了与Tyr889,TRP861和Ser904的氢键。这些相互作用表明ZINC43120769占据了与Olaparib相当的区域,与已知的关键残基相互作用,已知可以稳定结合口袋内的配体。化合物锌40934164与ARG878和ALA880形成氢键,而ZINC14584870形成了包括ARG878,ALA880,HIS862,SER864和SER904的强相互作用网络。具有较低结合能的化合物,锌14584870和锌43120769,与重要残基产生了更牢固的氢键和疏水相互作用,例如Arg878和Ser904,并在结合口袋中显示出更大的形状互补性。与几个活性部位残基的实质性相互作用强调了有效抑制PARP1活性的药物的能力。三种顶部化合物的氢键相互作用如图所示。 6。候选化合物的生理化学特性和化学结构在表中呈现4。表3对接得分,与化合物的氢键产生氢键的残基数量和残基的参考化合物Olaparib和候选化合物。图6

一个)复杂的锌40934164(b)复杂的锌43120769和(c)复杂的锌14584870。使用Pymol 3.0.4创建相互作用数字(https://www.pymol.org/),整个图是使用Microsoft Office Power Point组装的。
分子动力学模拟
进行了MD仿真分析以验证对接化合物-Parp1复合物的稳定性。实施这项研究将产生对配体和PARP1蛋白的动态行为的重要见解,以及蛋白质催化位点内化合物的稳定性。因此,对三个复合物和apo-parp1进行了100 ns全原子MD模拟。RMSD是确定MD模拟过程中蛋白质配合物的稳定性的重要参数。它决定了蛋白质原子运动与参考结构的平均差异。The RMSD of Apo PARP1 steadily increases over time, eventually reaching a plateau at 0.3 nm.This implies that the apo form of the protein undergoes conformational variations as the simulation proceeds, indicating flexibility in the absence of a ligand.The RMSD of PARP1 in association with ZINC43120769 is slightly lower than that of the apo form, indicating that this ligand helps to stabilize the protein structure.The RMSD values remain steady, ranging from 0.2 to 0.3 nm, indicating that the protein maintained overall structural stability during the simulation.ZINC40934164 compound has a similar RMSD trend as ZINC43120769, with somewhat higher values but still within a stable range (between 0.25 and 0.3 nm) as also evident from Fig. 7A. The consistent RMSD shows a stable interaction between ZINC40934164 and PARP1, albeit with slightly greater flexibility than ZINC43120769.ZINC14584870 compound has the most stable RMSD profile, with values consistently less than 0.2 nm throughout the simulation.This shows that ZINC14584870 formed the most stable complex with PARP1 among all the compounds thereby stabilizing the protein.All the complexes exhibit RMSD values lower then apo and reference compound Olaparib.

RMSD and RMSF analysis of the Apo, reference drug (Olaparib) and complexes ZINC43120769, ZINC40934164 and ZINC14584870.RMSD and RMSF plotted over the time of 100Â ns for all the simulated systems.
Second simulation run was performed to assess reproducibility of the complexes.In Fig. 8。RMSD plots compare the structural stability of the complexes across both runs.

RMSD analysis over 100Â ns for complexes (一个) ZINC43120769, (b) ZINC40934164 and (c) ZINC14584870.The plots compare the RMSD of first run and the second MD simulation run.First run black, and second run red line.
The results of the second simulation run are consistent with the first run.Throughout the 100Â ns simulation, complexes ZINC43120769 and ZINC14584870 exhibited excellent stability, maintaining RMSD values consistently around 0.2Â nm。For complex ZINC43120769, an initial fluctuation was observed during the first 50Â ns, after which the RMSD stabilized and closely followed the pattern of the first run, suggesting convergence towards a stable conformation.However complex ZINC40934164 exhibited greater fluctuations in the second run with RMSD values slightly higher than in the first run.Despite this variation, the deviations remained within an acceptable range less than 0.25Â nm, showing that the complex did not experience any significant conformational changes and maintained its overall structural integrity.Furthermore, the RMSD values of all the ligand-bound complexes remained lower than those of the apo form, highlighting the stabilizing effect of ligand binding.This further supports the structural integrity of the complexes over time.
Figure 7B depicts the RMSF values for the apo PARP1 and its complexes with the three compounds.RMSF calculates the average variation of each residue around its mean position during the simulation, providing information about the flexibility of specific residues of the protein.In this study, fluctuations were particularly observed in several loop regions of the protein, specifically around the amino acid residues Glu883- Thr887, Ser781-Asp788, Thr825-Ala828, and Thr910-Gln912.The maximum RMSF values recorded for Apo, ZINC43120769, ZINC40934164, and ZINC14584870 were 0.67 nm, 0.52 nm, 0.54 nm, and 0.45 nm, respectively.The active site residues remained stable for all the complexes (Table5)。The RMSF value for the Apo form of PARP1 form was the greatest, indicating a system with more flexibility than the others.The compound ZINC14584870 demonstrated the lowest maximum RMSF among the three, suggesting that it is more effective in stabilizing the protein structure.
Compactness of simulated systems
The Radius of Gyration (Rg) is an important metric for determining the structural compactness of PARP1 and compound complexes over the simulation period.To investigate the compactness of the simulation systems, radius of gyration analysis was carried out using “gmx-gyrate†command of GROMACS.This parameter outlines the global stability and folding pattern of protein and its ligand bounded forms.A higher value of Rg denotes less compactness, less stability and expanded protein state.In contrast, lower Rg value suggests a compact, stable and condensed protein configuration.The Rg profiles for protein complexes and the apo form were examined using a 100 ns MD simulation.The Rg values of the complexes ZINC40934164 and ZINC14584870 were found to be surprisingly stable, oscillating within a small range of 182–184 nm as can be seen in Fig. 9。A modest drop in Rg was found between 35 and 45 ns, indicating a tiny structural shift in the protein structure.However, after this change, the Rg values stabilized within the initial range, indicating that the overall compactness of the protein–ligand complexes remained constant for the rest of the simulation.In comparison, the compound ZINC43120769 showed slightly different behavior.Initially, the Rg values were consistent with the other compounds, ranging from 182–184 nm.However, after 40 ns, Rg gradually decreased, eventually reaching 179–180 nm.This drop indicates a modest shrinkage in the overall structure of the protein–ligand complex, which remained stable throughout the simulation.The apo version of the protein showed a significantly altered Rg profile.Unlike the complexes with ligands, the apo form exhibited constant Rg variations throughout the simulation, indicating a lack of structural stability and higher flexibility in the absence of a binding ligand.The mean Rg values of apo PARP1, ZINC43120769, ZINC40934164 and ZINC14584870 were 1.81 nm, 1.80 nm, 1.82 nm, and 1.82, respectively.

Radius of gyration of the Apo and complexes ZINC43120769, ZINC40934164 and ZINC14584870.plotted over the time of 100Â ns.
Principle component analysis (PCA)
PCA was used to analyze the conformational dynamics of the three ligand–protein complexes, ZINC43120769, ZINC40934164, and ZINC14584870.Figure 10PCA plots show the distribution of conformations collected during the simulation, indicating significant variations in each complex’s dynamic behavior.The distribution of points in each plot represents the conformational space explored by the protein during the simulation.Among the three complexes, ZINC43120769 (Fig. A) and ZINC14584870 had more compact and well-defined clusters, indicating greater structural stability.The PCA plot of ZINC43120769 shows two dominating conformational states with little dispersion.

Principle component analyses of the three complexes in a two-dimensional space defined by Principal Component 1 (PC1) and Principal Component 2 (PC2).((一个) ZINC43120769, (b) ZINC40934164 and (c) ZINC14584870.Similarly, ZINC14584870 exhibited a dense and continuous distribution, showing balanced structural, implying the lower degree of structural fluctuations.
In contrast, ZINC40934164 exhibited a ring-like shape with a central void, indicating a more dynamic and adaptable conformational landscape.This pattern shows frequent transitions between distinct states, as well as possible energy barriers that prevent the system from settling into a stable shape.Overall, PCA analysis suggests that ZINC43120769 and ZINC14584870 exhibit greater stability, while ZINC40934164 is more flexible and less likely to maintain a stable binding conformation.
MM-PBSA of the complexes
The MM-PBSA approach is an essential tool for understanding the energy aspects of molecular interactions after MD simulations.This is significant because it provides a precise and comprehensive picture of the binding free-energy landscape, revealing critical information about the stability and affinity of molecular compounds.Decomposing the total binding energy into distinct components such as van der Waals, electrostatic, and solvation energies allows for an accurate assessment of the attraction intensity of molecules.The MM-PBSA method was used to compute the binding free energies of PARP1 complexes with ZINC43120769, ZINC40934164, and ZINC14584870.The analysis included different energy terms such as ), electrostatic (ΔEelec), van der Waals (ΔEvdw), solvation (ΔGSOLV), gas-phase (ΔGGAS), and total binding free energies.The energy values of each energy term of all complexes are detailed in Table6。Table 6 MM-PBSA profile of PARP1 in complex with compounds ZINC43120769, ZINC40934164, and ZINC14584870.Electrostatic interactions were favorable for ZINC43120769 (− 21.08 kcal/mol) and ZINC14584870 (− 8.12 kcal/mol) but strongly unfavorable for ZINC40934164 (48.63 kcal/mol), indicating probable repulsion.
Among the three compounds ZINC14584870 has the most beneficial van der Waals interactions, followed by ZINC43120769 and ZINC40934164.The favorable van der Waals indicates strong hydrophobic interactions with residues like Trp861, Tyr896, and Ala880.Solvation energy in positive of ZINC43120769 and ZINC14584870 indicates that these compounds are more hydrophobic and prefers to remain in a less polar environment, like the protein binding site.Compound ZINC43120769 and ZINC14584870 have favorable gas-phase interactions, while ZINC40934164 had negative interactions with ΔGGAS 29.29 kcal/mol.Overall, ZINC43120769 and ZINC14584870 had the highest binding affinities (− 21.83 kcal/mol and − 20.38 kcal/mol, respectively which is comparable to the total binding energy of the reference drug Olaparib. While ZINC40934164 had the lowest (− 9.04 kcal/mol). These data indicate that ZINC43120769 and ZINC14584870 are the most potent binders to LasR, owing to strong van der Waals and electrostatic contacts, whereas ZINC40934164 poor binding is related to unfavorable gas-phase interactions, despite its favorable solvation energy. The Binding free energy results are consistent with the above RMSD, RMSF and PCA results.
Toxicity measurements
The toxicity of the compounds was predicted using ProTox-II.The proTox-II server determines the toxic properties of compounds through the predicted median lethal dose (LD50) in mg/kg weight.ZINC40934164 and ZINC43120769 were found to be in class IV while ZINC14584870 was in class V indicative of non-toxicity and non-irritating.As class IV indicate very low toxicity while class V indicate non-toxic This finding highlights these hits to be favorable in terms of toxicity.The predicted LD50 for ZINC40934164 was 760Â mg/kg, LD50 2500/kg for ZINC43120769,and for ZINC14584870 was 1500Â mg/kg.According to LD50 values the compound ZINC40934164 exhibit moderate toxicity while the other compounds exhibit no toxicity.The toxicity profile of the selected candidates is presented in Table7。Table 7 Toxicity measurements of the predicted compounds.Chemical diversity analysis
The 2D chemical structure of the known drugs (Fig.Â
11) and the selected compounds were compared in terms of their similarity.The TC, which ranges from 0 to 1, is a quantitative measure of molecular similarity based on structural fingerprints.The TC values close to 1 indicating more similarity between two molecules.Figure 12shows the TC values of the candidate compounds compared to the known PARP1 drugs, allowing for a direct comparison of TC values between each candidate molecule and each known inhibitors.This study found varied degrees of structural alignment with the known inhibitors, showing distinct similarities and contrasts among the candidates.This variation in similarity profiles underlines each candidate’s distinct structural features, supporting the concept that these compounds have different binding affinities and selectivities for PARP1.Physiochemical properties of the candidate compounds and the reported PARP1 drugs were also conducted (Fig. 13)。The properties compared include Molecular Weight (MolWT), LogP (Lipophilicity), H-Bond Donor/Acceptor counts, and Topological Polar Surface Area (TPSA).The selected compounds have physicochemical features similar to those of the four previously known PARP1 inhibitors.All compounds have molecular weights more than 400 g/mol, indicating that they are in a comparable size range, which may encourage binding affinity.The LogP values of the candidate compounds are approximately 4 which is comparable to Olaparib and Rucaparib.

Chemical structures (2D) of the known PARP drugs.

Tanimoto coefficient similarity between the candidate compounds and the reported drugs of PARP1 (Olaparib, Niraparib, Talazoparib, and Rucaparib).TC values ranges between 0 and 1. Values close to 1 indicates high similarity between two compounds.

Physiochemical Properties comparison between the selected compounds (ZINC14584870, ZINC4093164, and ZINC43120769) and the known inhibitors Olaparib, Niraparib, Talazoparib, and Rucaparib.((一个) Molecular weight (MolWT), (b) LogP, (c) Number of H-Bond donors and acceptors, (d) Topological Surface Area (TPSA).
讨论
In this study we employed machine learning based virtual screening to identify potential compounds as therapeutics for prostate cancer by targeting the PARP1 enzyme.PARP1 (Poly(ADP-ribose) polymerase 1) is a crucial enzyme implicated in DNA repair and has been implicated in cancer progression and resistance to chemotherapy.Inhibiting PARP1 has emerged as a promising strategy for cancer treatment, particularly in tumors with defective DNA repair mechanisms4,,,,5。The extensive dataset, encompassing 6510 active inhibitors and 287 decoy compounds, provided a solid foundation for machine learning-based predictive modeling.A library of 9000 phytochemicals was targeted to find promising inhibitors against PARP1.Machine learning model and drug likeness filtering finally yielded 40 active compounds for PARP.ZINC43120769, ZINC40934164, and ZINC14584870 were the three compounds that stood out after docking because of their substantial binding affinities toward the PARP1 active site.We compared these molecules to the well-known PARP1 inhibitor olaparib, which has crystallized with the PARP1 ART domain.According to our investigation, Arg878, His862, and Ser904, three important catalytic residues in the PARP1 enzyme, were created via hydrogen bonding by all three compounds.The validity of our findings was reinforced by the observation that these interactions aligned with the findings of previous studies16,,,,43,,,,44。The interaction profile of the selected candidate ligands was compare able to Olaparib.All candidate compounds form H-bonds with Arg878 which is critical for Olaparib activity.Among all ZINC14584870 showed a more diverse interaction profile, which may indicate its binding stability.To further confirm the potential of these substances, we employed MD simulations and MM-PBSA (Molecular Mechanics/Poisson-Boltzmann Surface Area) analyses.The MD simulations revealed that the chemicals ZINC43120769 and ZINC14584870 significantly stabilized the PARP1 structure over time.MD simulations of the protein–ligand complexes provided insights into the stability and dynamics of these interactions.The RMSD and RMSF analyses indicated that compounds ZINC14584870, ZINC40934164, and ZINC43120769 effectively stabilize the PARP1 protein structure, with ZINC14584870 showing the most stable interaction profile45。According to the RMSD values, among all ZINC14584870 maintained the most constant conformation which assess the stability of the protein–ligand complex.Furthermore, the MM-PBSA analysis demonstrated that these two compounds exhibited a high affinity for PARP1, indicating their potential as effective inhibitors.Overall, our findings show that machine learning-based virtual screening, paired with vast phytochemical libraries, can reveal promising novel PARP1 inhibitors.The compounds ZINC43120769 and ZINC14584870, in particular, show tremendous promise due to their binding affinity (− 21.83 kcal/mol and − 20.38 kcal/mol, respectively) and stabilizing actions on the PARP1 enzyme, making them excellent candidates for further research in prostate cancer therapy.Niraparib, Olaparib, Talazoparib, and Rucaparib are some of the most extensively investigated PARP1 inhibitors for PC46,,,,46,,,,47,,,,48,,,,49,,,,50。These inhibitors impede PARP1’s catalytic activity, resulting in DNA damage buildup and cell death in cancer cells.Talazoparib, has a high trapping potency on PARP-DNA complexes, which improves its effectiveness in treating BRCA-mutated malignancies.Olaparib, the first clinically authorized PARP1 inhibitor, has demonstrated remarkable effectiveness in BRCA-mutant PC and is being investigated for additional malignancies46,,,,47。Niraparib is remarkable for its wide range of applications in PC maintenance treatment48。Comparing the candidate compounds evaluated in this study revealed structural and interaction similarities, indicating potential efficacy in PARP1 inhibition.Notably, olaparib and talazoparib are known to exhibit strong binding interactions with catalytic residues, particularly Arg878 and Ser904 with PARP1.Docking results revealed that candidate compounds ZINC43120769 and ZINC14584870 also interact with these key residues, replicating Olaparib binding conformation.Furthermore, these candidate compounds have comparable molecular characteristics;such as molecular weights (MolWT) and topological polar surface areas (TPSA) roughly matching those of recognized inhibitors.The substantial structural similarity of ZINC14584870 to Talazoparib (Fig. 10) shows that this candidate compound may stabilize PARP1 through comparable processes, thereby increasing DNA damage and synthetic lethality in prostate cancer cells lacking DNA repair pathways.
结论
In this study we used machine learning based screening to identify and evaluate potential inhibitors for prostate cancer treatment.Different models like SVM, KNN, NB and RF were used to predict active inhibitors from a dataset of 6510 known compounds and 2871 decoys.The Random Forest model showed the highest accuracy, specificity, and AUC scores.It was used to screen 9510 phytochemicals, identifying 181 potential active compounds.Further analysis using Lipinski’s Rule of Five narrowed down the compounds to 40 drugs like compounds.Further molecular docking studies showed compounds ZINC14584870, ZINC43120769, and ZINC40934164 had strong binding interactions with the PARP1 active site residues including the crucial residues Arg878 and Ser904, similar to the reference inhibitor Olaparib, demonstrating high affinity and effective interaction.Overall MD simulations and MM-PBSA results demonstrated that two compounds ZINC14584870 and ZINC43120769.These compounds have a stable and robust contact with the target, making them ideal candidates for future research and therapeutic applications.Further experimental validation and clinical trials will be required to confirm their efficacy and therapeutic potential.
数据可用性
Data is provided within the manuscript or supplementary information files.
参考
Wong, M. C. et al.Global incidence and mortality for prostate cancer: Analysis of temporal patterns and trends in 36 countries.欧元。乌罗尔。 70, 862–874 (2016).
文章一个 PubMed一个 Google Scholar一个
Howrey, B. T., Kuo, Y.-F., Lin, Y.-L.& Goodwin, J. S. The impact of PSA screening on prostate cancer mortality and overdiagnosis of prostate cancer in the United States.J. Gerontol.Series A Biomed.科学。医学科学。 68, 56–61 (2013).
文章一个 Google Scholar一个
Sekhoacha, M. et al.Prostate cancer review: Genetics, diagnosis, treatment options, and alternative approaches.分子 27, 5730 (2022).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
D’Amours, D., Desnoyers, S., D’Silva, I. & Poirier, G. G. Poly (ADP-ribosyl) ation reactions in the regulation of nuclear functions.生物化学。J. 342, 249–268 (1999).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Swindall, A. F., Stanley, J. A. & Yang, E. S. PARP1: Friend or foe of DNA damage and repair in tumorigenesis?.癌症 5, 943–958 (2013).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Pascal, J. M. The comings and goings of PARP1 in response to DNA damage.DNA修复 71, 177–182 (2018).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Farmer, H. et al.Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy.自然 434, 917–921 (2005).
文章一个 广告一个 CAS一个 PubMed一个 Google Scholar一个
Mendes-Pereira, A. M. et al.Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors.embo mol。医学 1, 315–322 (2009).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
McCabe, N. et al.Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly (ADP-ribose) polymerase inhibition.能。res。 66, 8109–8115 (2006).
文章一个 CAS一个 Google Scholar一个
Thomas, C. & Tulin, A. V. Poly-ADP-ribose polymerase: Machinery for nuclear processes.摩尔。方面医学。 34, 1124–1137 (2013).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Weaver, A. N. & Yang, E. S. Beyond DNA repair: Additional functions of PARP1 in cancer.正面。Oncol。 3, 290 (2013).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Langelier, M.-F., Planck, J. L., Roy, S. & Pascal, J. M. Structural basis for DNA damage–dependent poly (ADP-ribosyl) ation by human PARP1.科学 336, 728–732 (2012).
文章一个 广告一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Hottiger, M. O., Hassa, P. O., Lüscher, B., Schüler, H. & Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases.趋势生物化学。科学。 35, 208–219 (2010).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Zong, C. et al.PARP mediated DNA damage response, genomic stability and immune responses.int。J.癌症 150, 1745–1759 (2022).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Thapa, K., Khan, H., Sharma, U., Grewal, A. K. & Singh, T. G. Poly (ADP-ribose) polymerase-1 as a promising drug target for neurodegenerative diseases.生命科学。 267, 118975 (2021).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Mateo, J. et al.A decade of clinical development of PARP inhibitors in perspective.安。Oncol。 30, 1437–1447 (2019).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Zhang, S.-H.等。Design, synthesis and biological evaluation of dual inhibitors targeting AR/AR-Vs and PARP1 in castration resistant prostate cancer therapy.生物。药物。 180, 117485 (2024).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Messina, C. et al.Combining PARP inhibitors and androgen receptor signalling inhibitors in metastatic prostate cancer: A quantitative synthesis and meta-analysis.欧元。乌罗尔。Oncol。 7, 179–188 (2024).
文章一个 PubMed一个 Google Scholar一个
Herencia-Ropero, A. et al.The PARP1 selective inhibitor saruparib (AZD5305) elicits potent and durable antitumor activity in patient-derived BRCA1/2-associated cancer models.Genome Medicine 16, 107 (2024).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Boussios, S. et al.Poly (ADP-Ribose) polymerase inhibitors: Talazoparib in ovarian cancer and beyond.Drugs in R 20, 55–73 (2020).
文章一个 CAS一个 Google Scholar一个
Deshmukh, D. & Qiu, Y. Role of PARP1 in prostate cancer.是。J. Clin。经验。乌罗尔。 3, 1 (2015).
CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Teyssonneau, D. et al.Prostate cancer and PARP inhibitors: progress and challenges.J. Hematol。Oncol。 14, 1–19 (2021).
文章一个 Google Scholar一个
Dara, S., Dhamercherla, S., Jadav, S. S., Babu, C. M. & Ahsan, M. J. Machine learning in drug discovery: A review.艺术品。Intell。修订版 55, 1947–1999 (2022).
文章一个 PubMed一个 Google Scholar一个
Kourou, K., Exarchos, T. P., Exarchos, K. P., Karamouzis, M. V. & Fotiadis, D. I. Machine learning applications in cancer prognosis and prediction.计算。结构。生物技术。J. 13, 8–17 (2015).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Zhang,H。等。An integrated deep learning and molecular dynamics simulation-based screening pipeline identifies inhibitors of a new cancer drug target TIPE2.正面。Pharmacol. 12, 772296 (2021).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Zhou, R. et al.Machine learning-aided discovery of T790M-mutant EGFR inhibitor CDDO-Me effectively suppresses non-small cell lung cancer growth.Cell Commun.信号。 22, 1–25 (2024).
文章一个 Google Scholar一个
Liu, T., Lin, Y., Wen, X., Jorissen, R. N. & Gilson, M. K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities.核酸res。 35, D198–D201 (2007).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Mysinger, M. M., Carchia, M., Irwin, J. J. & Shoichet, B. K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking.J. Med。化学 55, 6582–6594 (2012).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Karamizadeh, S. et al.An overview of principal component analysis.J. Signal Inf.过程。 4, 173–175 (2020).
Khamis, M. A., Gomaa, W. & Ahmed, W. F. Machine learning in computational docking.艺术品。Intell。医学 63, 135–152 (2015).
文章一个 PubMed一个 Google Scholar一个
Ballester, P. J. & Mitchell, J. B. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking.生物信息学 26, 1169–1175 (2010).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Karthikeyan, M., Vyas, R., Karthikeyan, M. & Vyas, R. Machine learning methods in chemoinformatics for drug discovery.实践。Chemoinf.133–194 (2014).
Trott, O. & Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading.J. Comput。化学 31, 455–461 (2010).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Bekker, H. et al。在4th International Conference on Computational Physics((PC 92)。252–256 (World Scientific Publishing).Best, R. B. et al.
Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone Ï•, ψ and side-chain χ1 and χ2 dihedral angles.J. Chem。Theory Comput. 8, 3257–3273 (2012).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Wang, E. et al.End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design.化学修订版 119, 9478–9508 (2019).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Case, D. A. et al.Amber 2021(University of California, 2021).
Banerjee, P., Eckert, A. O., Schrey, A. K. & Preissner, R. J. ProTox-II: A webserver for the prediction of toxicity of chemicals.核酸res。 46, W257–W263 (2018).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Pollastri, M. P. Overview on the rule of five.Curr。Protoc.Pharmacol. 49, 9–12 (2010).
文章一个 Google Scholar一个
Plinski, E. F. & Plinska, S. Veber’s rules in terahertz light.(2020)。
Priya, N. & Shobana, G. Application of machine learning models in drug discovery: A review.int。J. Emerg。技术。 10, 268–275 (2019).
CAS一个 Google Scholar一个
Cano, G. et al.Automatic selection of molecular descriptors using random forest: Application to drug discovery.专家系统。应用。 72, 151–159 (2017).
文章一个 Google Scholar一个
Dawicki-McKenna, J. M. et al.PARP1 activation requires local unfolding of an autoinhibitory domain.摩尔。细胞 60, 755–768 (2015).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Langelier, M.-F., Zandarashvili, L., Aguiar, P. M., Black, B. E. & Pascal, J. M. NAD+ analog reveals PARP1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains.纳特。社区。 9, 844 (2018).
文章一个 广告一个 PubMed一个 PubMed Central一个 Google Scholar一个
Adelakun, N. et al.Discovery of new promising USP14 inhibitors: Computational evaluation of the thumb-palm pocket.J. Biomol.结构。Dyn. 40, 3060–3070 (2022).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Agarwal, N. et al.Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: TALAPRO-2 phase III study design.未来的Oncol。 18, 425–436 (2022).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Smith, M. R. et al.Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): A multicentre, open-label, phase 2 trial.柳叶刀Oncol。 23, 362–373 (2022).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
de Bono, J. et al.Olaparib for metastatic castration-resistant prostate cancer.N. Engl。J. Med。 382, 2091–2102 (2020).
文章一个 PubMed一个 Google Scholar一个
Abida, W. et al.Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration.J. Clin。Oncol。 38, 3763–3772 (2020).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Anscher, M. S. et al.FDA approval summary: Rucaparib for the treatment of patients with deleterious BRCA-mutated metastatic castrate-resistant prostate cancer.肿瘤学家 26, 139–146 (2021).
文章一个 CAS一个 PubMed一个 Google Scholar一个
致谢
The authors extend their appreciation to the Researchers Supporting Project number (RSP2025R506), King Saud University, Riyadh, Saudi Arabia for funding this work.
竞争利益
作者没有宣称没有竞争利益。
附加信息
Publisher’s note
关于已发表的地图和机构隶属关系中的管辖权主张,Springer自然仍然是中立的。
Electronic supplementary material
以下是电子补充材料的链接。
权利和权限
开放访问
本文在Creative Commons Attribution-Noncormercial-Noderivatives 4.0国际许可下获得许可,该许可允许任何非商业用途,共享,分发和复制以任何媒介或格式的形式,只要您提供适当的原始作者和来源的信用,请符合原始作者和来源,并提供了与Creative Commons的链接,并指示您是否修改了许可的材料。您没有根据本许可证的许可来共享本文或部分内容的改编材料。The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.要查看此许可证的副本,请访问http://creativecommons.org/licenses/by-nc-nd/4.0/。Reprints and permissions
引用本文

Aldakheel, F.M., Alduraywish, S.A. & Dabwan, K.H.
Integrating machine learning driven virtual screening and molecular dynamics simulations to identify potential inhibitors targeting PARP1 against prostate cancer.Sci代表 15, 12764 (2025).https://doi.org/10.1038/s41598-025-97208-8
已收到:
公认:
出版:
doi:https://doi.org/10.1038/s41598-025-97208-8