
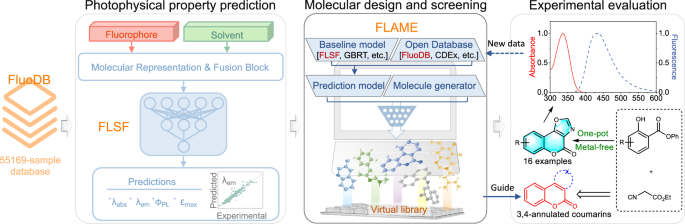
A modular artificial intelligence framework to facilitate fluorophore design
作者:Qian, Linghui
Introduction
Luminescent molecules have found widespread applications in numerous fields1,2,3, among which fluorophores have attracted increasing attention in bioimaging due to their small size, chemical tractability, and low cost4,5. To meet specific requirements such as the penetration depth and detection sensitivity in bioimaging, the underlying structure-property relationship (SPR) of fluorophores is important for designing compounds with proper excitation wavelength and desired brightness6,7,8. However, our knowledge of this relationship remains limited9,10,11,12,13, which is largely due to two reasons: (1) Data sparsity. That is, all possible structural modifications should be tested to illustrate the SPR of a specific fluorophore, but met with synthetic challenges. Moreover, the problem also lies in the limited access to complete, comparable, and meaningful photophysical data of existing fluorophores14. (2) Multiple interrelated factors may affect the fluorescence. A subtle modification in the structure may lead to significant optical changes, and the fluorescence may further be influenced by the surrounding environment, leaving the rational design of fluorophores difficult6,8,15.
Recently, machine learning-based data-driven science has shown tremendous potential to become a very useful tool across various disciplines16,17,18, such as predicting molecular properties19, virtual screening20, and molecular generation21. In the case of fluorescence, multiple intricately intertwined properties need to be considered for the molecular design, including maximum absorption wavelength—λabs, maximum emission wavelength—λem, photoluminescence quantum yield—ΦPL, and molar absorption coefficient—εmax6,15. Pioneered by Tsuda et al., a massively parallelized version of de novo molecule generator (ChemTS) was employed to design fluorophores with absorption/emission wavelengths and oscillator strengths calculated by quantum chemical computation, generating 3643 candidate fluorophores by using 1024 cores for 5 days22. Though powerful, the high computational cost must be considered.
Due to its end-to-end paradigm, machine learning can learn directly from the data to identify implicit patterns and make predictions without any prior knowledge, promising for fluorescence prediction. For instance, Ju et al. established a database (ChemFluor) recording the optical properties of over 4300 solvated fluorophores23. Both the fluorophore and solvent were characterized using molecular descriptors or fingerprints, which were combined as the input for predicting photophysical parameters using the Gradient Boosted Regression Trees (GBRT) model. Similarly, Park’s group developed a graph convolutional network (GCN)-based model24 and employed the integrated gradients method25 sequentially to predict seven optical properties and obtain attributions of atoms/functional groups/solvents to the optical properties. Very recently, Tsai et al. modified the SchNet model to introduce the solvation embedding outside the interaction layers so as not to overly amplify the solute-solvent interaction and provided enhanced prediction for ΔEabs and ΔEemi26.
As researchers in fluorescent probes27,28,29, we’re keen to make an easy-operation toolkit that allows the generation of structure-new fluorophores with desired optical performance efficiently, to explore the frontiers of fluorophores with a minimized burden on chemical synthesis and experimental tests30.
Very recently, Park et al. developed a generative deep learning (Gen-DL) model to generate molecules with seven predefined optical properties31. Alternatively, to fully exploit the chemical space, optical property prediction models can be introduced to molecule generators for efficient sampling to select optimal structures with desired optical properties.
Herein, we systematically compiled experimental data to build a new fluorophore database named FluoDB (Fig. 1), consisting of 55,169 fluorophore-solvent pairs, as the machine learning algorithm asks for large volumes of data to acquire effective information. Compared with existing databases, FluoDB improves in both data volume and molecular diversity, categorized with 16 core fluorescent scaffolds and 728 subgroups. Then we proposed a new prediction model, FLSF (FLuorescence prediction with fluoroScaFfold-driven model), in which a domain-knowledge-derived fingerprint encoded by 728 fluorescent-scaffold subgroups (called fluoroscaffold) is fused to traditional message passing neural networks (MPNN; reported to outperform SchNet, DTNN, and the Transformer in predicting UV-Vis spectra32) using the gated recurrent unit (GRU). In benchmarking tests, FLSF is advantageous at quickly and accurately predicting optical properties over previous state-of-the-art (SOTA) models. Its reliability and potential were further validated through a series of interpretability analyses. To guide the fluorophore design directly, we set up an artificial intelligence (AI) framework, FLAME (FLuorophore design Acceleration ModulE), by integrating different open-source databases, prediction models, and molecule generators. Using Reinvent 433 as a representative molecule generator, a series of compounds with predicted properties were generated. Among them, 3,4-oxazole-fused coumarins were synthesized using a novel one-pot synthetic methodology, giving an unreported compound with bright fluorescence, and exhibiting the potential of FLAME in accelerating fluorophore design.

Overview of the user-friendly framework, FLAME, assembled from the latest databases (including FluoDB, the database constructed in the current study), prediction models (i.e., FLSF (FLuorescence prediction with fluoroScaFfold-driven model) constructed in the current study together with other state-of-the-art prediction models), and molecule generators, together with its application in the design of unreported fluorophores with desired fluorescence followed by experimental evaluation.
Results
Data collection and processing
In our previous study, a database (SMFluo1) focusing on near infrared fluorophores was constructed, containing five widely used fluorescent scaffolds29. The limitation in data volume of SMFluo1 makes it difficult to meet the requirements of deep learning algorithms, particularly those based on graph neural networks (GNN), including GCN and Attentive FP34, leading to moderately good prediction accuracy. In addition, to evaluate a fluorophore for bioimaging, four key photophysical parameters (λabs, λem, ΦPL, and εmax) are needed, where λabs and λem are related to the penetration depth and ΦPL×εmax indicates the brightness15. Of note, other factors including the blinking, thermal stability, photobleaching, and labeling specificity should be taken into consideration when designing probes for bioimaging, but parameters for these properties were not included in FluoDB due to limited access6. Taking these into consideration, data collection was carried out as follows (Fig. 2a): (i) Literature survey via searching the name of fluorescent scaffolds on PubMed; (ii) Retrieval of experimental data from various open-source databases23,35,36,37,38,39,40 and supplement with four photophysical parameters & solvent information from the original literature. These data were processed after the combination (see “Data processing” in Methods).

a General pipeline for data collection and processing to construct the new database, FluoDB, followed by systematic analysis and statistics of FluoDB to visualize the optical properties of various fluorophores in different solvents. b UMAP (Uniform Manifold Approximation and Projection) of different databases (Deep4Chem36, DyeAgg38, ChemFluor23, ChemDataExtrator (CDEx)35, and SMFluo129) using Morgan fingerprints. The UMAP algorithm was applied with a neighborhood size of 10 and a minimum distance of 0.3. The number of unique compounds in each database is listed in the bracket. c Distribution of various fluorescent scaffolds in different databases. Source data are provided as a Source data file.
Most fluorescent compounds are derived from some basic scaffolds and they may share common optical characteristics; thus, we categorized the fluorophores into twelve classic fluorescent scaffolds and four non-classical scaffolds (Fig. S1; detailed skeletal structures for 728 subgroups are shown in Table S1). FluoDB, a new database containing 35,528 unique fluorophores and 55,169 fluorophore-solvent pairs, was therefore constructed with SMILES of fluorophores/solvents, category of fluorescent scaffolds, experimental photophysical data, and original reference.
Compared to representative open-source databases (e.g., Deep4Chem36, DyeAgg38, ChemFluor23, ChemDataExtrator (CDEx)35, and SMFluo129), FluoDB gets improved in the number of molecules and the richness in optical information (Fig. 2b and Fig. S2), exhibiting much higher molecular diversity according to the data distribution and structural analysis (Fig. S3 and Tables S2–3). In addition, different scaffolds distribute relatively even in both FluoDB and Deep4Chem, while the data of each category is largely enriched in FluoDB (Fig. 2c and Table S4).
Data analysis with FluoDB
With FluoDB, the correlations between different parameters were investigated (Fig. S4) and indicated an obvious positive correlation between λabs and λem. In addition, molecular weight (MW) also has a certain positive correlation with λabs, λem, and εmax, which is consistent with the scatter plot analysis (Fig. S5) and experimental results (i.e., introducing large π bridging moieties or strong electron acceptors/donors is commonly used for longer absorption and emission wavelengths41, and these modifications often increase the MW). External factors such as the surrounding solvent are reported to influence the optical properties of certain fluorophores42. To investigate this in a large scope, fluorophores with experimental data available for different solvents (≥5) were selected from FluoDB. The variance distribution of each photophysical parameter for selected molecules in different solvents is shown in Fig. S6, where these parameters do vary along the change of solvent type, underscoring the importance of solvents when predicting optical properties.
As mentioned earlier, we divided the fluorescent scaffolds into 16 types (Fig. S1 and Table S1), and any input fluorophore can be quickly classified accordingly to explore potential commonalities from the same group. As indicated in Figs. S7–22, a discrepancy was found in λabs and λem distribution with different scaffolds, where most groups centered in the UV-Vis range, while BODIPY, porphyrin, and squaraine lie in longer wavelength (above 550 nm)43,44. In addition, larger wavelength tunability was found from acridine, naphthalimide, coumarin, and cyanine. Besides, Δλ (Stokes shift, Δλ = λem − λabs) of BODIPY, porphyrin, and squaraine is relatively small (~25 nm). Statistical analysis of such large-scale data is indicative for choosing the ideal fluorescent scaffold to start with, and we also prepared a toolkit where users can search for fluorophores with desired similarity as the molecule of interest from the database.
Workflow and prediction performance of FLSF
With FluoDB, open-source prediction models, including GBRT23, SMFluo29, UVVisML45, SchNet26, and ABT-MPNN46, were tested. Data in FluoDB-Lite (SMILES in the mixture/complex form were removed from FluoDB) was divided randomly in a ratio of 7:1:2 for training, validation, and testing, respectively (Table S5). As shown in Table 1 and Table S6, ABT-MPNN, a general molecular property prediction model based on an atom-bond Transformer, performed best in predicting λabs and λem, highlighting the advantage of combining Transformers with GNN for molecular representation. However, the introduction of attention mechanisms in ABT-MPNN led to 10 times slower training than UVVisML (a Directed MPNN, D-MPNN), despite the improved MAE for λabs and λem by 9.18% and 4.86%, respectively. For practicability, it is desirable to replace the attention mechanism in ABT-MPNN with a new way to speed up the training while maintaining the prediction accuracy.
As all fluorophores in FluoDB were classified into 16 core scaffolds and distribution discrepancy in optical properties among them was observed (Figs. S7–21), a special molecular fingerprint—fluoroscaffold (a 728-dimensional digital fingerprint encoded by 728 fluorescent-scaffold subgroups listed in Table S1), fused with the current feature extraction method based on MPNN, was designed for better molecular representation of fluorophores. A new prediction model (FLSF) was constructed based on it (Fig. 3a). As shown in Table 2, FLSF predicted well for different fluorophores, especially for BODIPY-based compounds (the largest proportion in FluoDB) with MAE of 6.44 nm/7.37 nm for λabs/λem. For non-classical scaffolds (i.e., [6 + 5], [6 + 6], 6-n-5, 6-n-6), FLSF also has a good performance, promising for dealing with novel fluorophores. Overall, FLSF performs well at predicting λabs and λem (R2 = 0.94) and needs improvement at ΦPL and εmax (R2 ≈ 0.6; Fig. 3b). Then we conducted benchmark tests of FLSF and summarized the results in Table 1 and Table S6 for direct comparison with reported SOTA models. Obvious improvements were seen in the prediction accuracy of λabs, λem, and εmax by FLSF than ABT-MPNN (the same of ΦPL), at a much faster speed, indicating its great potential for high-throughput screening of candidate fluorophores. To check whether FLSF can capture the solvent effect, a multi-solvent test set (fluorophores with experimental data available for ≥4 different solvents) together with the control test set (fluorophores in the same solvent) was selected and the prediction performance of FLSF was compared with other baseline models. As shown in Tables S7–8, FLSF has the best prediction performance on the multi-solvent test set. To our delight, FLSF can also predict λabs and λem of fluorophores showing solvatochromism with high accuracy (Tables S9–10), further supporting its potency to capture solvent effects.

a The model architecture of FLSF. A domain-knowledge-derived fingerprint based on the 728 fluorescent-scaffold subgroups (called fluoroscaffold) is fused with a message-passing neural network (MPNN) for the feature extraction of the input fluorophore. The feature extraction of the solvent molecule is based on MPNN. The feature vectors of both the fluorophore and the solvent are input together to output a prediction of the property of interest. MLP: multilayer perceptron. b The overall prediction performance of FLSF for different photophysical parameters. λabs: maximum absorption wavelength; λem: maximum emission wavelength; ΦPL: photoluminescence quantum yield; εmax: molar absorption coefficient. c Comparison between FLSF (red points) and TD-DFT (time-dependent density functional theory) calculations (gray points) for λabs (left) and λem (right) prediction. MAE mean absolute error, R2 the coefficient of determination. Source data are provided as a Source data file.
Time-dependent density functional theory (TD-DFT) used to be the most widely used tool for predicting optical properties47. However, such traditional theoretical calculations require high computational and time costs48, faced with insufficient accuracy in predicting λabs and λem, much less in parameters like ΦPL involved in various radiation and non-radiation processes49. For direct comparison with FLSF, we collected 162 fluorophore-solvent pairs from FluoDB (Table S11) and used TD-DFT to calculate their λabs and λem (Fig. 3c and Table S12). The MAE of FLSF decreased by more than 0.2 eV for predicting λabs and λem than TD-DFT. Of note, FLSF can provide all prediction results in less than one second, while the average calculation time of TD-DFT exceeds 200 CPU hours in the current test set.
Interpretability analysis of FLSF
The interpretability of a model, illustrating how it makes decisions and achieves related results, helps to verify the reliability of the model and excavate valuable information from the data. First, we analyzed the interpretability of FLSF from the molecule-level perspective50. The embedding vectors from three states of FLSF were studied, namely, the state only treated by D-MPNN without fluoroscaffold integration, the state with fluoroscaffold integration but before solvent incorporation, and the state after solvent incorporation. According to the 2D-PCA (two-dimensional principal component analysis) dimension reduction distribution diagram (Fig. 4a), there is a clear difference in the distribution between short-wavelength and long-wavelength fluorophores in λabs and λem prediction tasks, indicating that FLSF can effectively identify their structural features with different wavelengths. Interestingly, the integration of fluoroscaffold makes this difference more significant, and the data distribution dispersion is further improved after the introduction of solvent, highlighting the importance of scaffold information for the prediction and implying that FLSF is sensitive in capturing subtle differences caused by solvents.

a FLSF embedding interpretability through PCA (Principal Component Analysis). 2D-PCA plots of the molecular embeddings at different stages: (Left) before integrating fluoroscaffold information, (Center) after integrating fluoroscaffold information, and (Right) after further integration of solvent information. Each dot is colored by experimental values. PC1: Principal Component 1; PC2: Principal Component 2. The intensity of the color scale represents the magnitude of the experimental values of the molecular parameters, with darker colors indicating higher experimental values. Source data are provided as a Source data file. λabs: maximum absorption wavelength; λem: maximum emission wavelength; ΦPL: photoluminescence quantum yield; εmax: molar absorption coefficient. b Summary of reported structural modification strategies for coumarin-based fluorophores (left) and atomic contributions learned by FLSF (right). The color bar values represent the normalized difference in the predicted values before and after masking specific atoms. EDG electron-donating group, EWG electron-withdrawing group, Exp. experimental, Pred. predicted.
Subsequently, the explicability analysis of FLSF at the atom-level perspective was conducted50. To be specific, each atom in the fluorophore was masked, and the prediction values (e.g., λem) before and after masking were compared to reveal the attribution of each atom. Coumarin was taken as the example since it has been derived to cover a wide range of wavelengths, providing invaluable SPR information (Fig. 4b, left) for validating the reliability of FLSF51,52. With a classic D-π-A structure, the introduction of electron-donating groups (EDG) on the phenyl ring and electron-withdrawing groups (EWG) on the lactone ring can effectively achieve redshift of coumarin according to experimental experience. Representative examples in Fig. 4b (right) demonstrate that FLSF has grasped such rules. In addition, researchers found that the replacement of ketone with imine at position 2 can also produce redshift53 (e.g., compound e-g), and FLSF has also mastered it. Of note, although coumarin derivatives with substitution other than oxygen at position 1 are not recorded in FluoDB, FLSF can indicate the contribution of oxygen at this position to the redshift, which is also supported by recent experimental results54. It implies that FLSF has good generalization ability/reliability and may provide new structural modification suggestions for fluorophore design.
Construction of FLAME for fluorophore design
While large databases and various property prediction models have significantly advanced our knowledge of the SPR of certain molecules, a gap exists in their direct applications for molecular design. Therefore, we aimed to build a multifunctional software package, FLAME, to meet the practical needs of researchers for novel fluorophore design by integrating the database, prediction models, and molecule generators into one framework (Fig. 5a). FLAME provides six open-source fluorophore databases, including FluoDB (Fig. 5b). Users can input the molecule of interest to search for related information of existing molecules in the database, as well as to train the model with different databases for illustrating the data impact. Meanwhile, FLAME offers six open-source prediction models (i.e., FLSF, UVVisML, ABT-MPNN, SchNet, SMFluo, and GBRT), which can be combined with the above datasets to meet various requirements from different users. In-parallel comparison between different combinations also helps to identify the best settings for specific parameter prediction (Table 3).

a The framework of FLAME to facilitate the fluorophore design. FLAME, assembled from the latest databases and prediction models, is feasible for various applications, including virtual screening, molecular generation, and structural optimization. SMILES: Simplified Molecular Input Line Entry System; MLP: multilayer perceptron; λabs: maximum absorption wavelength; λem: maximum emission wavelength; ΦPL: photoluminescence quantum yield; εmax: molar absorption coefficient. b The basic workflow of FLAME for various applications, including database search, photophysical property prediction, and creating unreported molecules with predicted optical properties by integrating different fluorophore databases, prediction models, and molecule generators. Databases including Deep4Chem36, DyeAgg38, ChemFluor23, CDEx35, SMFluo129, and our FluoDB; prediction models including previously reported UVVisML45, ABT-MPNN46, SchNet26, SMFluo29, GBRT23, and our FLSF (FLuorescence prediction with fluoroScaFfold-driven model).
To provide structure-new compounds with predicted optical properties directly, a newly reported open-source generative AI framework, Reinvent 433, was introduced. As a scoring tool embedded in FLAME for molecular design, the speed of training and predicting is critical for the prediction model. FLSF proven good at these two aspects was coupled with Reinvent 4 herein (users can make their own choice). With the help of FLAME, both de novo molecular generation and structural modifications can be achieved. For example, if users are interested in the development of novel BODIPY derivatives, they can set the desired photophysical parameters (single or multiple parameters) with FLAME. Then, newly generated molecules (not recorded in FLAME’s built-in database) belonging to BODIPY with predicted properties will be screened out. Alternatively, users can input a parent structure of interest with desired parameters into FLAME to obtain optimized structures. Of note, both processes used to be highly dependent on specialized knowledge and years of experience, while FLAME is promising to think out of the box and offer fluorophore candidates more efficiently.
Experimental evaluation
With increasing interest in coumarin-based fluorescent probes due to their excellent biocompatibility, good structural flexibility, and tunable fluorescence52, FLAME is employed to guide the development of novel coumarin derivatives for concept proof (Fig. 6a and Fig. S23). Four optical parameters (λabs, λem, ΦPL, and εmax) were set as scoring targets. We trained the generative model and sampled one million molecules, with a focus on coumarin-type compounds during screening. From the virtual library generated by FLAME, 3,4-oxazole-fused coumarins attracted our attention due to their structural novelty and synthesizability. A variety of oxazole-containing dyes were reported to possess attractive photophysical properties, such as high fluorescence quantum yields55,56,57, while the fluorescence properties of 3,4-oxazole-fused coumarins have not been reported yet.

a Generation of unreported heterocyclic-fused coumarins with predicted optical properties by FLAME. FLSF: FLuorescence prediction with fluoroScaFfold-driven model; Reinvent 433: a newly reported open-source generative AI framework. b The new strategy for one-pot synthesis of 3,4-oxazole-fused coumarins. c Absorption (up) and emission (down) spectra of 3h and 3o recorded in different solvents. H2O: water; DMSO: dimethyl sulfoxide; EtOH: ethanol; DCM: dichloromethane. Source data are provided as a Source data file. d Confocal fluorescence images of living HeLa cells treated with different concentrations of 3o. The cell imaging was performed three times with similar results.
Available strategies for the synthesis of this scaffold include (a) heating of 7-N,N-dimethylamino-4-hydroxycoumarin in the presence of nitromethane and DABCO58, (b) synthesis from 4-hydroxy-3-nitrocoumarin and benzyl alcohol under gold nanoparticle or FeCl3 catalysis59, and (c) synthesis from 4-hydroxy-3-nitrocoumarin and acids in the presence of triphenylphosphine and phosphorus pentoxide under microwave irradiation60 (Fig. S24). The lack of structural diversity on the phenyl ring using reported strategies, together with the demand for simple and efficient synthetic procedures to construct diverse 3,4-oxazole-fused coumarins from readily available starting materials, drives us to develop new synthetic methodology. Inspired by our previous work in isocyanide chemistry61,62,63, we proposed a one-pot approach to synthesize 3,4-oxazole-fused coumarins from ethyl isocyanoacetates and phenyl salicylates promoted by base (Fig. 6b and Fig. S25). Under the optimized conditions (Table S13), 16 oxazole-fused coumarins were synthesized successfully (Figs. S26–27), carrying electron-donating or electron-withdrawing substituents on the phenyl ring. With these compounds in hand, their optical properties were evaluated (Figs. S28–29). Consistent with the prediction result from FLSF, the introduction of an amino group at the 6- or 7-position of the coumarin scaffold (i.e., 3h, 3o) led to a redshift in λabs and an increase in ΦPL (Table S14). Then, solvent effects on these two fluorophores were investigated (Fig. 6c and Table S15). As expected, the solvent polarity has a significant impact on their absorption/emission wavelengths, and 3o liberated much stronger emission than 3h in all solvents, which was selected for bioimaging. Brilliant fluorescence was observed in HeLa cells after 30-min incubation (Fig. 6d), indicating its potential for live-cell imaging.
Discussion
FLAME, a modular AI-assisted framework for fluorophore design, was developed to help researchers design de novo molecules with desired optical performance efficiently. To achieve this, we expanded the available fluorophore database by supplementing data from various aspects, such as fluorescent scaffold types and photophysical parameters, to give the biggest open-source fluorophore database to date, FluoDB, which contains 55,169 solvated fluorophores and 109,054 data entries including four key photophysical parameters, λabs, λem, ΦPL, and εmax. Compared with reported databases, FluoDB exhibits higher molecular diversity and data volume. By conducting a series of data analyses on FluoDB, insights were gained into the correlation between different photophysical parameters, as well as their relationships with molecular weight and solvent type.
To meet the requirement of scoring for molecular generation, the prediction model needs to be accurate and fast. FLSF with a domain-knowledge-derived fingerprint for characterizing fluorescent scaffolds (called fluoroscaffold: a 728-dimensional digital fingerprint) was designed and exhibited encouraging accuracy with a training speed 10 times faster than ABT-MPNN. In addition, FLSF’s predictive power was tested through a series of molecule-level and atom-level interpretability analyses. The attribution of each atom learned by FLSF is highly consistent with expertise. Based on FLSF, Reinvent 4 as a molecule generator was employed for de novo creation of fluorophore candidates. A series of 3,4-oxazole-fused coumarins yet-to-be-developed for fluorophores were synthesized using our newly developed metal-free approach via base-promoted tandem reaction of phenyl salicylates with isocyanoacetates. The predicted optical performance of these compounds is highly consistent with the experimental results (MAE = 13.3 nm for λabs, 0.093 for ΦPL, and 0.430 for log10εmax), and an unreported coumarin derivative with brilliant fluorescence (ΦPL = 0.541, log10εmax = 4.314 in water) promising for bioimaging was obtained.
The above results exemplify the advance of FLAME in facilitating the design of new fluorophores, which can reduce the burden on trial-and-error experiments by simply inputting the desired photophysical parameters into the black box of FLAME. Multi-step computational processes can thus be executed automatically and can be handled by anyone without a prerequisite for expertise in either fluorescence or computation. With its modular architecture, FLAME can be further updated with new data/algorithms to advance with time in accelerating fluorophore development. Moreover, synthetic accessibility predicting models64,65,66,67,68 can be further integrated into FLAME for synthesizability scoring during the sampling, and retrosynthetic analysis tools (e.g., AiZynthFinder69, Retro*70, and ASKCOS71) can also be incorporated into FLAME which can help with retrosynthesis planning towards the fluorophore candidate, making fluorophore design and synthesis more efficiently.
Methods
Data collection
In addition to searching literature via PubMed by using fluorescent scaffolds as keywords, we also compiled and supplemented multiple open-source databases, including Deep4Chem36, ChemFluor23, Dye Aggregation (DyeAgg)38, ChemDataExtrator (CDEx)35, DYES39, PhotochemCAD40, and Dye-Sensitized Solar Cell Database (DSSCDB)37. FluoDB is currently available for the experimental photophysical data, including maximum absorption wavelength (λabs), maximum emission wavelength (λem), photoluminescence quantum yield (ΦPL), and molar absorption coefficient (εmax), which are key factors in photochemical studies. During the data collection, if multiple peaks were found in the absorption/emission spectra for the same fluorophore-solvent pair, the peak with the longest wavelength/largest intensity was collected for λabs and λem. The majority of the experimental values were obtained at 298 K, so the effect of temperature was not considered in the model development.
Data processing
First, we removed the invalid data: (1) remove data without solvent information or with gas as the solvent; (2) remove data whose SMILES cannot be converted to valid chemical structures. Then we did some general processing to limit the data range of each photophysical parameter: (1) remove data with ΦPL above 1; (2) remove data with λabs or λem below 200 nm or above 1500 nm; (3) remove data with εmax above ten million. During the redundant data processing, we set difference thresholds for each parameter. The difference threshold is 5 nm for λabs and λem, 0.1 for ΦPL, and 0.02 for log10εmax. For each fluorophore-solvent pair, redundant data from different resources were removed if exceeding the difference threshold, and the average value of the remaining data was put into the database.
Finally, all fluorophores were standardized using SMILES notation, and a dictionary mapping to convert solvent names and acronyms into SMILES was constructed. Furthermore, the number of solvent types was streamlined from 393 to 72 by removing those that occurred less than 10 times. Since these less-used solvents account for a small portion of the original data (~2000 entries), their removal will not affect the data diversity.
Model training and evaluation
We trained and tested four optical property parameters separately. Data containing fluorophores in the mixture/complex form (containing water, metal ions, etc. in the SMILES) were removed from FluoDB (6309 entries were removed) to give FluoDB-Lite (the original FluoDB is also named FluoDB-Full to differentiate it from FluoDB-Lite when applicable), before a random split with a ratio of 7:1:2 (detailed in Table S5). Of note, some implausible data (εmax < 100) were removed from the test set during εmax prediction (Table S6). The hyperparameters for FLSF were tuned by Bayesian optimization on the validation set. All regression models are evaluated by MAE (mean absolute error), MSE (mean-square error), and RMSE (root-mean-square error). The training was conducted on two servers—OCHPC and SYHPC. The OCHPC server has 2 Intel Skylake Gold 6132 processors and 192GB RAM, along with an NVIDIA Tesla K80 24GB GPU. The SYHPC server has 4 Intel 8360H processors, 3TB RAM, and an NVIDIA A100-40GB GPU. The hyperparameters for FLSF are shown in Table S16.
TD-DFT calculations
The molecules used for the TD-DFT (time-dependent density functional theory) tests were sourced from the previously divided test set. We selected data that contained all four parameters, excluded molecules containing ions, and restricted the number of heavy atoms to less than 30. Initial geometries were refined using semi-empirical tight-binding density functional theory (GFN2-xTB) followed by geometry optimizations at the B3LYP/6-31 + G(d)/IEFPCM level of theory in the Gaussian 16 software package. TD-DFT calculations72 were performed with CAM-B3LYP/6-31 + G(d)/IEFPCM level of theory.
General information for chemical synthesis
1H, 13C, 19F NMR spectra were recorded using JNM-ECZ 400S (400 MHz) spectrometer. Chemical shifts were reported in parts per million (ppm), and the residual solvent peak was used as an internal reference: 1H (chloroform δ 7.26; DMSO δ 2.50), 13C (chloroform δ 77.16; DMSO δ 39.52). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, dd = doublet of doublets, ddd = doublet of doublet of doublets), coupling constants (Hz) and integration. Melting point (MP) was obtained on Buchi M-560. For thin layer chromatography (TLC), Huanghai TLC plates (HSGF 254) were used, and compounds were visualized with a UV light at 254 nm. High-resolution mass spectra (HRMS) were obtained on an Agilent G6545 spectrometer using an electron spray ionization time-of-flight (ESI-TOF) source. Unless otherwise noted, all reactions were carried out under an ambient atmosphere; exclusion of air or moisture was not required. Anhydrous and deuterated solvents were purchased from commercial suppliers and used as received without further purification. Phenyl salicylates 1a-1p (Fig. S26) were prepared according to literature73. Ethyl isocyanoacetate (2) was purchased from commercial suppliers and used without further purification.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets and prediction results are available at Figshare (https://doi.org/10.6084/m9.figshare.26317933)74. All data generated in this study are provided in the Source data file. Source data are provided with this paper.
References
Chen, Y., Wang, S. & Zhang, F. Near-infrared luminescence high-contrast in vivo biomedical imaging. Nat. Rev. Bioeng. 1, 60–78 (2023).
Bryden, M. A. & Zysman-Colman, E. Organic thermally activated delayed fluorescence (TADF) compounds used in photocatalysis. Chem. Soc. Rev. 50, 7587–7680 (2021).
Kumar, R. et al. Revisiting fluorescent calixarenes: from molecular sensors to smart materials. Chem. Rev. 119, 9657–9721 (2019).
Wang, K. et al. Fluorescence image-guided tumour surgery. Nat. Rev. Bioeng. 1, 161–179 (2023).
Wu, L., Huang, J., Pu, K. & James, T. D. Dual-locked spectroscopic probes for sensing and therapy. Nat. Rev. Chem. 5, 406–421 (2021).
Grimm, J. & Lavis, L. D. Caveat fluorophore: an insiders’ guide to small-molecule fluorescent labels. Nat. Methods 19, 149–158 (2022).
Hong, G., Antaris, A. L. & Dai, H. Near-infrared fluorophores for biomedical imaging. Nat. Biomed. Eng. 1, 0010 (2017).
Remmel, A. How to keep the lights on: the mission to make more photostable fluorophores. Nature 630, 258–260 (2024).
Wang, S. et al. Anti-quenching NIR-II molecular fluorophores for in vivo high-contrast imaging and pH sensing. Nat. Commun. 10, 1058 (2019).
Wang, C. et al. Twisted intramolecular charge transfer (TICT) and twists beyond TICT: from mechanisms to rational designs of bright and sensitive fluorophores. Chem. Soc. Rev. 50, 12656 (2021).
Yan, K. et al. Ultra-photostable small-molecule dyes facilitate near-infrared biophotonics. Nat. Commun. 15, 2593 (2024).
Zhou, J., Ren, T.-B. & Yuan, L. The strategy to improve the brightness of organic small-molecule fluorescent dyes for imaging. Chin. Chem. Lett. https://doi.org/10.1016/j.cclet.2024.110644 (2024).
Lovell, T. C., Branchaud, B. P. & Jasti, R. An organic chemist’s guide to fluorophores–understanding common and newer non-planar fluorescent molecules for biological applications. Eur. J. Org. Chem. 27, e202301196 (2024).
Cavazos-Elizondo, D. & Aguirre-Soto, A. Photophysical properties of fluorescent labels: a meta-analysis to guide probe selection amidst challenges with available data. Anal. Sens. 2, e202200004 (2022).
Jiang, G. et al. Chemical approaches to optimize the properties of organic fluorophores for imaging and sensing. Angew. Chem. Int. Ed. 63, e202315217 (2024).
Puszkarska, A. M. et al. Machine learning designs new GCGR/GLP-1R dual agonists with enhanced biological potency. Nat. Chem. 16, 1436–1444 (2024).
Reid, J. P. & Sigman, M. S. Holistic prediction of enantioselectivity in asymmetric catalysis. Nature 571, 343–348 (2019).
Du, Y. et al. Machine learning-aided generative molecular design. Nat. Mach. Intell. 6, 589–604 (2024).
Lewis, L. et al. Improved machine learning algorithm for predicting ground state properties. Nat. Commun. 15, 895 (2024).
Gentile, F. et al. Artificial intelligence-enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 17, 672–697 (2022).
Zhang, O. et al. ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling. Nat. Mach. Intell. 5, 1020–1030 (2023).
Sumita, M. et al. De novo creation of a naked eye-detectable fluorescent molecule based on quantum chemical computation and machine learning. Sci. Adv. 8, eabj3906 (2022).
Ju, C. W., Bai, H., Li, B. & Liu, R. Machine learning enables highly accurate predictions of photophysical properties of organic fluorescent materials: emission wavelengths and quantum yields. J. Chem. Inf. Model. 61, 1053–1065 (2021).
Joung, J. F. et al. Deep learning optical spectroscopy based on experimental database: potential applications to molecular design. JACS Au 1, 427–438 (2021).
Joung, J. F., Han, M., Jeong, M. & Park, S. Beyond Woodward–Fieser rules: design principles of property-oriented chromophores based on explainable deep learning optical spectroscopy. J. Chem. Inf. Model. 62, 2933–2942 (2022).
Hung, S.-H., Ye, Z.-R., Cheng, C.-F., Chen, B. & Tsai, M.-K. Enhanced predictions for the experimental photophysical data using the featurized Schnet-bondstep approach. J. Chem. Theory Comput. 19, 4559–4567 (2023).
Qian, L., Li, L. & Yao, S. Q. Two-photon small molecule enzymatic probes. Acc. Chem. Res. 49, 626–634 (2016).
Wang, W. et al. Real-time imaging of cell-surface proteins with antibody-based fluorogenic probes. Chem. Sci. 12, 13477–13482 (2021).
Shao, J. et al. Prediction of maximum absorption wavelength using deep neural networks. J. Chem. Inf. Model. 62, 1368–1375 (2022).
Koscher, B. A. et al. Autonomous, multi-property-driven molecular discovery: From predictions to measurements and back. Science 382, eadi1407 (2023).
Han, M., Joung, J. F., Jeong, M., Choi, D. H. & Park, S. Generative deep learning-based efficient design of organic molecules with tailored properties. ACS Cent. Sci. https://doi.org/10.1021/acscentsci.4c00656 (2024).
McNaughton, A. D. et al. Machine learning models for predicting molecular UV−Vis spectra with quantum mechanical properties. J. Chem. Inf. Model. 63, 1462–1471 (2023).
Loeffler, H. H. et al. Reinvent 4: Modern AI-driven generative molecule design. J. Cheminform. 16, 20 (2024).
Xiong, Z. et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 63, 8749–8760 (2020).
Beard, E. J., Sivaraman, G., Vazquez-Mayagoitia, A., Vishwanath, V. & Cole, J. M. Comparative dataset of experimental and computational attributes of UV/Vis absorption spectra. Sci. Data 6, 307 (2019).
Joung, J. F., Han, M., Jeong, M. & Park, S. Experimental database of optical properties of organic compounds. Sci. Data 7, 295 (2020).
Venkatraman, V., Raju, R., Oikonomopoulos, S. P. & Alsberg, B. K. The dye-sensitized solar cell database. J. Cheminform. 10, 18 (2018).
Venkatraman, V. & Chellappan, L. K. An open access data set highlighting aggregation of dyes on metal oxides. Data 5, 45 (2020).
Ksenofontov, A. A., Lukanov, M. M. & Bocharov, P. S. Can machine learning methods accurately predict the molar absorption coefficient of different classes of dyes? Spectrochim. Acta A Mol. Biomol. Spectrosc. 279, 121442 (2022).
Taniguchi, M. & Lindsey, J. S. Database of absorption and fluorescence spectra of >300 common compounds for use in PhotochemCAD. Photochem. Photobiol. 94, 290–327 (2018).
Wang, S., Li, B. & Zhang, F. Molecular fluorophores for deep-tissue bioimaging. ACS Cent. Sci. 6, 1302–1316 (2020).
Klymchenko, A. S. Solvatochromic and fluorogenic dyes as environment-sensitive probes: design and biological applications. Acc. Chem. Res. 50, 366–375 (2017).
Resch-Genger, U., Grabolle, M., Cavaliere-Jaricot, S., Nitschke, R. & Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 5, 763–775 (2008).
Würth, C., Geißler, D., Behnke, T., Kaiser, M. & Resch-Genger, U. Critical review of the determination of photoluminescence quantum yields of luminescent reporters. Anal. Bioanal. Chem. 407, 59–78 (2015).
Greenman, K. P., Green, W. H. & Gomez-Bombarelli, R. UVVisML (0.0.2). Zenodo https://doi.org/10.5281/zenodo.5986671 (2022).
Liu, C., Sun, Y., Davis, R., Cardona, S. T. & Hu, P. ABT-MPNN: an atom-bond transformer-based message-passing neural network for molecular property prediction. J. Cheminform. 15, 29 (2023).
Adamo, C. & Jacquemin, D. The calculations of excited-state properties with time-dependent density functional theory. Chem. Soc. Rev. 42, 845–856 (2013).
Rubesova, M., Muchova, E. & Slavicek, P. Optimal tuning of range-separated hybrids for solvated molecules with time-dependent density functional theory. J. Chem. Theory Comput. 13, 4972–4983 (2017).
Charaf-Eddin, A., Le Guennic, B. & Jacquemin, D. Excited-states of BODIPY–cyanines: ultimate TD-DFT challenges? RSC Adv. 4, 49449–49456 (2014).
Wu, J. et al. ALipSol: An attention-driven mixture-of-experts model for lipophilicity and solubility prediction. J. Chem. Inf. Model. 62, 5975–5987 (2022).
Sharma, S. J. & Sekar, N. Deep-red/NIR emitting coumarin derivatives—synthesis, photophysical properties, and biological applications. Dyes Pigm. 202, 110306 (2022).
Cao, D. et al. Coumarin-based small-molecule fluorescent chemosensors. Chem. Rev. 119, 10403–10519 (2019).
Rabahi, A. et al. Synthesis and optical properties of coumarins and iminocoumarins: Estimation of ground- and excited-state dipole moments from a solvatochromic shift and theoretical methods. J. Mol. Liq. 195, 240–247 (2014).
Matikonda, S. S., Ivanic, J., Gomez, M., Hammersley, G. & Schnermann, M. J. Core remodeling leads to long wavelength fluoro-coumarins. Chem. Sci. 11, 7302–7307 (2020).
Takechi, H., Oda, Y., Nishizono, N., Oda, K. & Machida, M. Screening search for organic fluorophores: syntheses and fluorescence properties of 3-azolyl-7-diethylaminocoumarin derivatives. Chem. Pharm. Bull. 48, 1702–1710 (2000).
Mahuteau-Betzer, F. & Piguel, S. Synthesis and evaluation of photophysical properties of series of π-conjugated oxazole dyes. Tetrahedron Lett. 54, 3188–3193 (2013).
Xing, Z.-H. et al. Novel oxazole-based emitters for high efficiency fluorescent OLEDs: synthesis, characterization, and optoelectronic properties. Tetrahedron 73, 2036–2042 (2017).
Satyanarayana, I., Manjappa, K. B. & Yang, D.-Y. Nitromethane as a surrogate cyanating agent: 7-N,N-dimethylamino-4-hydroxycoumarin-catalyzed, metal-free synthesis of α-iminonitriles. Green. Chem. 22, 8316–8322 (2020).
Vlachou, E.-E. N., Armatas, G. S. & Litinas, K. E. Synthesis of fused oxazolocoumarins from o-hydroxynitrocoumarins and benzyl alcohol under gold nanoparticles or FeCl3 catalysis. J. Heterocycl. Chem. 54, 2447–2453 (2017).
Balalas, T. D. et al. One-pot synthesis of 2-substituted 4H-chromeno[3,4-d]oxazol-4-ones from 4-hydroxy-3-nitrocoumarin and acids in the presence of triphenylphosphine and phosphorus pentoxide under microwave irradiation. SynOpen 2, 105–113 (2018).
Qian, L. et al. Catalytic atroposelective dynamic kinetic resolution of biaryl lactones with activated isocyanides. Org. Lett. 23, 5086–5091 (2021).
Tao, L.-F. et al. Diastereo- and enantioselective silver-catalyzed [3 + 3] cycloaddition and kinetic resolution of azomethine imines with activated isocyanides. Angew. Chem. Int. Ed. 61, e202202679 (2022).
Luo, Z.-H. et al. Torsional strain-independent catalytic enantioselective synthesis of biaryl atropisomers. Angew. Chem. Int. Ed. 61, e202211303 (2022).
Yu, J. et al. Organic compound synthetic accessibility prediction based on the graph attention mechanism. J. Chem. Inf. Model. 62, 2973–2986 (2022).
Chen, S. & Jung, Y. Estimating the synthetic accessibility of molecules with building block and reaction-aware SAScore. J. Cheminform. 16, 83 (2024).
Seo, S., Lim, J. & Kim, W. Y. Molecular generative model via retrosynthetically prepared chemical building block assembly. Adv. Sci. 10, 2206674 (2023).
Gao, W., Mercado, R. & Coley, C. W. Amortized tree generation for bottom-up synthesis planning and synthesizable molecular design. In The Tenth International Conference on Learning Representations. (ICLR, 2022).
Guo, J. & Schwaller, P. Directly optimizing for synthesizability in generative molecular design using retrosynthesis models. Chem. Sci. https://doi.org/10.1039/d5sc01476j (2025).
Saigiridharan, L. et al. AiZynthFinder 4.0: developments based on learnings from 3 years of industrial application. J. Cheminform. 16, 57 (2024).
Chen, B., Li, C., Dai, H. & Song, L. Retro*: learning retrosynthetic planning with neural guided A* search. https://doi.org/10.48550/arXiv.2006.15820 (2024).
Tu, Z. et al. ASKCOS: an open source software suite for synthesis planning. Preprint at https://arxiv.org/abs/2501.01835 (2025).
Runge, E. & Gross, E. K. U. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 52, 997–1000 (1984).
Serratore, N. A. et al. Integrating metal-catalyzed C-H and C-O functionalization to achieve sterically controlled regioselectivity in arene acylation. J. Am. Chem. Soc. 140, 10025–10033 (2018).
Zhu, Y. et al. A modular artificial intelligence framework to facilitate fluorophore design. Figshare https://doi.org/10.6084/m9.figshare.26317933 (2025).
Zhu, Y. ChemloverYuchen/FLAME: FLAME-1.0. Zenodo https://doi.org/10.5281/zenodo.14842448 (2025).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82473881 and 82273887 to L.Q.), the Joint Funds of the Zhejiang Provincial Natural Science Foundation of China (LTZ22B020001 to J.L.), Key Research and Development Program of Zhejiang Province (2025C02087 to Z.M.), and Zhejiang University. We also thank Prof. Tingjun Hou for providing the server for computation and Prof. Chang-Yu Hsieh for helpful discussions throughout this work. The computational calculations were also supported by the HPC Center of Zhejiang University (Zhoushan Campus) and the High-performance Computing Platform of YZBSTCACC.
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, Y., Fang, J., Ahmed, S.A.H. et al. A modular artificial intelligence framework to facilitate fluorophore design. Nat Commun 16, 3598 (2025). https://doi.org/10.1038/s41467-025-58881-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58881-5