
AI设计的,抗突变的宽性中和抗体针对多个SARS-COV-2菌株
作者:Pan, Lurong
介绍
SARS-COV-2的新菌株的演变使许多先前批准的抗体疗法无效,尤其是针对病毒尖峰蛋白的抗体疗法。在其基因组中的数百种突变中。峰值蛋白的ACE2受体结合结构域(RBD)内的人是研究人员的主要重点,因为这些突变会严重影响峰值蛋白与ACE2受体的结合强度。例如,在B.1.427和B.1.429中发现的L452R替代物可显着降低病毒对Bamlanivimab的敏感性1,以及适度降低其对Bamlanivimab和Etesevimab组合的敏感性1,,,,2,,,,3。在本研究中,我们使用人工智能(AI)来生成更多\({10}^{9} \)
在硅酸盐中的抗体突变,然后实际上筛选了可以广泛结合并与已知的尖峰蛋白RBD变体结合的候选抗体序列。图神经网络(GNNS)4
是专门针对图形数据而设计的神经网络体系结构。图中的节点旨在学习包含有关其相关邻居的信息的嵌入。嵌入可以用适用于节点标记,边缘预测和图形表示的特征功能,并具有正确的读数和汇总方法5,,,,6,,,,7,,,,8。GNN的内在设计使其非常适合研究分子和生物学相互作用以及其他化学和物理特性。因此,我们试图以基于图的方式描述抗体 - 抗原相互作用。
基于语言的网络还可以基于以下假设来建模蛋白质,即主要蛋白质结构类似于自然语言序列9,,,,10,,,,11,,,,12,,,,13,,,,14。氨基酸之间的隐藏依赖性和相互作用可以通过在基本复发性神经网络中固有设计的时间动态训练,例如长期短期存储网络(LSTM)和变形金刚神经网络。
我们探索了GNN和自然语言处理架构的几种建模策略,其中分别使用基于图的和基于语言的表示来描述蛋白质序列。
总体而言,这项研究描述了一种基于AI的方法使用深度学习,该方法可以使用蛋白质序列捕获抗体 - 抗原结合的信息,而无需任何其他数据。该模型可以预测蛋白质蛋白相互作用对快速发展的靶标(例如SARS-COV-2的不同菌株)的突变影响。我们的研究描述了使用深度学习模型来计算设计有效和广谱突变,以针对病毒尖峰蛋白的各种菌株进行,随后进行了湿与LAB实验证实了这一发现。由于这种AL驱动的抗体发现方法的有效性,因此可以在未来的大流行中快速发现治疗剂。这种方法还为设计常规蛋白药物发现的设计开辟了新的门,该大门可以广泛地与多种抗原结合或具有优化的选择性。
方法
通过AI的硅抗体亲和力成熟建模
培训和测试数据集
我们开发了基于深神经网络的抗体亲和力成熟模型。我们检查了以下公开策划的数据集以进行模型开发:Skempi数据库15抗体结合(AB结构)数据库16,观察到的抗体空间17和Uniprot18。在Skempi和AB界面上验证了所得模型的性能,这两者都策划了具有相关PDB结构的蛋白质 - 蛋白质复合物以及单位和多站点突变。在此数据策划过程中,仅将来自该蛋白质蛋白质复合物的序列数据用于模型训练。AB界代表自由能的变化突变变体的结合亲密关系(g)的结合)\(\ text {kcal}/\ text {mol} \),而Skempi为野生型复合物和突变体的亲和力提供了实验解离常数(KD)值。
深度学习模型
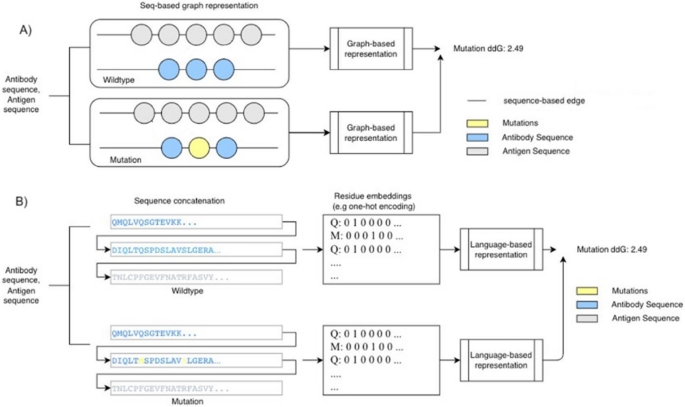
这项研究旨在检查是否仅使用主要序列输入来预测抗体 - 抗原复合物亲和力。因此,为了降低计算成本并将模型应用扩展到蛋白质(尚未解决的结构),我们探索了一个模型,该模型仅需要蛋白质的氨基酸序列作为预测结合亲和力的输入。图 1说明了建模策略的两个示例。

开发了两种建模方法的设计,以支持基于序列的抗体亲和力设计。((一个)图形神经网络表示亲和力培训(b)基于语言的(BILSTM或变压器)神经网络表示亲和力培训。
在我们的研究中,设计和实施了两种不同的方法来预测绑定,即图表和基于语言的表示。基于图的表示形式利用基于异质聚合图神经网络体系结构的神经网络19。这将使GNN通过汇总多个读取层来形成图表的清晰表示。GNN用于这种基于序列的方法,因为它们将淋巴结信息传播到邻居节点19允许局部蛋白质序列特征由模型表示。对于基于语言的表示,考虑了两个不同的体系结构,即双向LSTM(BILSTM)模型和变压器模型。选择BilstM表示蛋白质序列,因为蛋白质的主要结构不一定与蛋白质的3D形状相关。因此,在对蛋白质序列进行建模时,向前和向后的上下文都很重要。变压器模型20使用大量数据使用诸如uniref50的数据库中使用大量数据的验证模型的嵌入(矢量表示)21。
分类任务可以预测突变抗体和给定抗原之间的结合亲和力变化。我们认为突变导致这两种突变(即\(\ delta \ delta {\ text {g}} <0 \))或减少(即\(\ delta \ delta {\ text {g}}> 0 \))分别将亲和力作为正样本或负样本。我们的模型旨在提取氨基酸的潜在特性,这些特性最佳地用于最佳地描绘“增强”和弱化的粘合剂之间。
对于GNN模型,将基于GNN层之间异质聚集的模型用作主要体系结构。图形数据通常包含一些信息,作为节点和节点之间的连接作为边缘。在这种情况下,每个氨基酸残基被编码为代表一个节点的数值值,边缘是节点之间的连接。使用5幅卷积层对该GNN进行了训练,并使用每个读数层中使用金字塔功能堆叠。将所有图层的读数汇总在一起,并在所有块中添加密集的连接。此外,在每个折叠的左5O(L5O)方法之后,使用最多500个时期对该模型进行了训练。使用SGD优化器训练每个时期,学习率为0.02,重量衰减为0.0001。
在语言模型中,序列数据以不同的方式用于蛋白质序列表示。首先,在BilstM模型中,单个氨基酸残基被转换为数值一式壁编码表示。该表示形式以与蛋白质序列相同的顺序进行编码\(20 * n \)其中n表示蛋白质序列的最大长度。比该长度短的序列用零填充。该表示被输入到3 LSTM层和随后的致密层中。同样,该模型的最多为500个时期,每个折叠的左5O(L5O)方法。使用SGD优化器训练每个时期,学习率为0.02。接下来,使用Protbert和ESM2模型,从默认预验证的模型中编码嵌入式20参数。然后将其直接传递到密集的层中,并使用SGD优化器进行培训,学习率为0.02,500个时期。
模型评估
我们使用基于蛋白质家族区分的分布跨验证评估了模型的准确性和可伸缩性。我们的验证策略采用了一种离开5-out-out-out-out-out-out-un-of-5o方法,其中五个蛋白质家族及其相关的复合物(野生型和突变)被随机固定为验证集,而其余数据则用作训练集。该设计确保模型在测试过程中遇到了完全新颖的序列模式。
从我们包含60个独特蛋白质家族的策划数据集中,每个验证折叠都保留了5个随机选择的蛋白质家族,保证验证蛋白仍然完全分布训练数据(图S1)。为了确认这种抽样方法的公正性质,我们使用成对的全球比对百分比得分进行了所有60个蛋白质家族的全面相似性分析。任何两个蛋白质家族之间观察到的最大相似性仅为74%,大多数分数明显降低。鉴于我们数据集中的平均蛋白质序列超过500个残基,即使是最相似的家族也有大约130个氨基酸残基,从而在训练和测试集之间建立了真正的分布条件。随后将所得训练的模型完善并集成到Sentinusai®(我们的专用,无结构的大分子平台)中。
实际上,Sentinusai®的内在亲和力成熟过程通过系统地在全面的虚拟突变体虚拟库中系统地搜索高亲和力的粘合剂来模拟自然免疫系统。这种计算方法可以探索躯体突变空间,极大地超过了任何湿实验室方法的能力。通过优化的工程和分布式计算策略,我们实现了比传统方法快10,000倍的计算效率,从而可以快速识别最佳抗体候选物,否则这些抗体候选者否则就需要进行过多的广泛实验筛选。
用于识别COVID-19中和抗体的计算工作流程
数据收集
我们从Gisaid中检索了1300多种不同的历史SARS-COV-2菌株(包括Wildtype(B.1)和Delta)22,,,,23,,,,24截至2021年8月26日,数据库。选择了三种野生型抗体,CR3022,Casirivimab(Regen 10,933)和Imdevimab(Regen 10987)作为模板中的模板,以用于硅交叉结合抗体设计。CR3022抗体25是一种对SARS-COV-1特异性的单克隆抗体,是从人类康复血浆中获得的,该患者是从严重急性呼吸综合征(SARS-COV-1)中恢复的,这是一种与导致COVID-19的新型冠状病毒密切相关的病毒。CR3022与新型冠状病毒交叉反应,尽管其结合亲和力不足以中和病毒并阻止其感染细胞25,,,,26,,,,27,,,,28。casirivimab和imdevimab都29,是Regeneron开发的单克隆抗体鸡尾酒;他们针对Wildtype和Delta菌株的功能得到了证实。
在计算机突变库中
我们的中硅突变库是通过仅考虑模板抗体副群上的突变来构建的。根据PDB库中的模板抗体CR3022的复合物中的SARS-COV-2 RBD的晶体结构映射了抗体招聘量[PDB ID:6W41]。仅当已知和使用PDB结构时才完成。否则,使用算法(例如Anarci)绘制抗体副膜,以识别重链和轻链中的CDR1/2/3区域30。在两个氨基酸侧链中心之间的5.5-â截止距离内鉴定了抗体和抗原之间的界面接触。然后,我们在副链和轻链上详尽地产生了单点突变,形成了抗体体细胞突变文库(图。 2:步骤1)。该策略从每个抗体模板(CR3022,Regen 10933和Remen 10987)产生了109多个突变体。

SARS-COV-2的基于AI基于AI的广泛中和抗体设计。
步骤1:SARS-COV-2交叉结合序列选择和病毒突变数据策展。步骤2:基于AI的抗体结合预测和对未来变体潜在候选序列的跨变相结合选择。步骤3:使用基于ELISA的测定法测量抗体的结合能力;并使用中和和细胞疗法(CPE)还原测定法测量抗体中和能力。
在Silico图书馆一代
第一轮中硅抗体设计的目的是发现具有改善结合亲和力的抗体,包括1300多种不同的历史SARS-COV-2菌株,包括Wildtype(B.1)和Delta菌株。因此,使用训练有素的机器学习模型,根据高跨结合得分选择抗体序列。这些亲和力得分(在诱变文库中的每种突变抗体与历史SARS-COV-2菌株中的每个独特的突变体S1蛋白之间)使用病毒S1蛋白序列作为抗原计算其VH和VL链。该分数代表了给定抗体对靶抗原的亲和力改善的可能性。为每个抗体 - 抗原对生成一个分数。对于每个SARS-COV-2峰值蛋白突变体,从109个体细胞突变空间中选择了最高得分最高的抗体作为该特定菌株的强粘合剂。然后,所有1300个SARS-COV-2菌株(包括B. 1和delta)按照上述方案计算并选择所有强粘合剂,如图2的步骤2所述。 2在此文库中的所有抗体序列中对64个不同的抗原序列进行评分,最终得分是不同抗原结合亲和力得分的平均值。SARS-COV-2变体在S1蛋白的不同区域发展并获取突变。
因此,我们假设可以与所有观察到的S1蛋白结合的抗体可能能够结合未来的S1变体。从所有1300种变体的常见强结构抗体候选物中。从中,我们选择了所有变体中最高平均预测分数的前50个跨结合候选者。此步骤结束了我们在Omicron之前对跨结合抗体的第一轮计算。
我们在2022年2月进行了第二轮计算,以进一步提高对Omicron的AB亲和力。遵循上面详述的相同过程,并选择了最高平均预测分数的前20个跨结合抗体序列。这是我们在Omicron之前对交叉结合抗体的第一轮计算。这一轮计算改善了对Omicron的AB亲和力。遵循上面详述的相同过程,并选择了最高平均预测分数的前20个跨结合抗体序列。
湿实验室实验
HEK293抗体生产,ELISA结合测试是在Sino Biological Inc(1400 Liberty Ridge Drive,Suite 101,Wayne,Wayne,PA 19087 USA)进行的。南方研究所(2000 9th Ave S,伯明翰,AL 35294)进行了冠状病毒细胞毒性测定。Elisa
我们测量了使用ELISA结合不同SARS-COV-2菌株(B.1,Delta,Omicron)RBD的每种抗体的能力。
ELISA使用涂层缓冲液(500毫升),其中包含\(\ text {cbs} \ left(0.75 \,\ text {g} \ right。\)) 和\({\ text {naco}} _ {3}/{\ text {nahco}} _ {3}(1.46 \)g)在a\(\ text {phof} 9.6 \)。B.1,Delta和Omicron菌株的每个RBD蛋白都以浓度制备\(0.03 \)和\(1 \ mu \ text {g}/\ text {ml} \), 进而\(100 \ mu \ text {l} \)将抗体的含量添加到每个孔中。板覆盖\(4 {}^{\ circ} \ text {c} \)过夜。涂层后,通过摇动和喘气去除去溶液。然后将井与\(2 \ text {\%} \)BSA。将蛋白质在室温下孵育1 h。然后将溶液丢弃,并用两次蛋白质洗涤\(300 \ mu \ text {l} \)洗脱缓冲液(PBS缓冲液含有2%Tween-\(200,\ text {ph} \)7.2 7.4)并拍打干燥。
每种抗体被稀释至\(1 \ mu \ text {g}/\ text {ml} \)带有装饰缓冲液的\(0.1 \ text {\%bsa} \)。\(\ text {} \的一百个microx \)然后将稀释的抗体添加到先前涂有RBD蛋白的每个井中。将溶液均匀混合,并在室温下孵育2 h。将溶液丢弃,并用三次洗涤抗体\(300 \ mu \)洗脱缓冲液和拍打干燥的。\(\ text {一百} \ mu \)杰克逊(Jackson):将山羊抗人IgG(Hâ+l)/HRP二抗添加到每种孔,混合均匀,并在室温下孵育1 h。然后将溶液丢弃,并用\(300 \ mu \)洗脱缓冲液和拍打干燥的。TMB基板解决方案\(一个\)和\(b \)以1:1的比率均匀混合\(200 \ mu \)将混合物的含量添加到每个孔中,并在室温下在黑暗的房间中孵育。通过添加来阻止反应\(50 \ mu \)在每个孔到每个孔的2米硫酸,然后在450 nm处立即测量每个孔中的吸光度。总体而言,我们从\(0.03 1 \ text {ug}/\ text {ml} \)RBD(WT,Delta,Omicron)蛋白和生成的单克隆抗体的7个系列稀释液生成IC50值。
冠状病毒细胞毒性测定
病毒通过劫持细胞机制复制,导致细胞死亡;但是,抗病毒药物可以阻止这种作用。我们使用CPE还原测定法来确定中和抗体是否改善了细胞活力。该测定是用表达ACE2受体的VERO E6细胞进行的,该细胞介导病毒感染。
在补充的最小必需培养基(MEM)中培养细胞\(10 \ text {\%} \)热灭活的胎牛血清(HI FBS)。CPE和毒性测定使用的细胞从MEM补充中收集的细胞\(1 \ text {\%} \)青霉素链霉素谷氨酰胺和\(2 \ text {\%hi fbs} \)然后重悬于每毫升200,000个细胞。将二十微升的细胞悬浮液(约4000个细胞)添加到每个孔中。
通过将固定数量的病毒颗粒与抗体的系列稀释液混合,然后进行CPE测定,检测到病毒的中和。我们添加了\(5 \ mu \)血清浸润抗体以及\(5 \ mu \)l在384孔板的每个孔中包含1000 TCID的病毒。将盘子孵育1 h\({37}^{\ circ} \ text {c} \),然后通过添加CPE测定\(20 \ mu \)细胞悬浮液。空白对照仅由细胞组成,而病毒对照不含抗体。盘子在\(\ text {in} 5 \ text {\%} \)公司272 H和90%的湿度。将Promega细胞滴度电动发光细胞活力测定试剂盒的30微升添加到每个孔中,然后在室温下孵育10分钟。使用Perkin Elmer Invision或BMG ClarioStar微板读取器读取发光,以测量细胞活力。将来自每个井的原始数据标准化为100%(无抗体)的抑制率,使用以下公式将CPE抑制%抑制%:
$$ \ text {抑制率}(\ text {\%})= 100 \ times \ frac {\ text {(test valie} - \ text {avg virus test va} \ text va} \ text {lue holly holly holly})}
CPE分析是在生物安全3级实验室中使用板盖上清晰盖之前的板进行的。
使用与CPE分析中使用的相同培养基串行稀释的抗体检测到抗体细胞毒性。由\(20 \ mu \ text {l} \)细胞和\(10 \ mu \ text {l} \)在多孔板中将抗体添加到每个孔中。含有细胞的井仅作为空白对照,而用氯化苯甲酸处理的细胞(井)(\(100 \ mu \ text {m} \)最终浓度)用作阴性对照。如CPE分析中所述读取发光。
结果
AL建模基准
我们研究了仅基于序列输入的两种不同的蛋白质序列建模方法,以提高抗体成熟的预测准确性。这些建模方法,如图所示 1,包括基于图的方法和基于语言的方法。指标是根据五倍得分平均的feave-5淘汰(L5O)方法计算的。以前未知样本的性能一直是深度学习神经网络最常见的挑战之一。我们的建模旨在在执行抗体亲和力预测的情况下改善模型的鲁棒性。我们研究了几种神经网络建模方法,以找到用于抗体筛选的优化模型,如表所示1。通过评估先前看不见的样本的预测准确性来评估每种方法的性能。
分类的性能通过ROC面积(AUC)评估。使用Pearson和Spearman相关系数评估了实验亲和力变化与预测值之间的相关性。
我们还根据Spearman排名系数比较了不同基于学习的方法的排名能力(表1),来自Protbert或ESM的令牌机20变压器模型用于从我们的训练数据中编码序列。基于图的模型(Spearmanâ= 0.54)优于基于语言的方法,但是观察到基于变压器的模型具有强大的嵌入方式,可以从几乎相等的水平上捕获蛋白质序列的有效数据。此外,我们评估了模型的预测性能在所示的几个未知复合物上(图。 3)。

使用分布外测试集的四个代表性复合物(野生型和突变)的模型预测概率的散点图。独立测试集中的每个目标的相似性得分相对于彼此的相似性得分小于0.21。X轴代表了G值,该值与每个样品的亲和力强度呈正相关,而Y轴代表模型预测的概率。相关性分别通过皮尔逊的相关值和Spearman的相关值来量化。
桌子1使用基于语言的模型和基于图形的模型显示了对改进的二进制分类和弱化的粘合剂的平均AUC。基准研究16列出以进行比较。
基于图的(AUC = 0.82)和基于语言的(AUC = 0.73)建模方法在区分增强和弱的粘合剂方面的表现优于或与发现工作室的表现更好或可比16,这是一种基于商业结构的非机械学习方法。与Discovery Studio不同,该工作室采用了源自主要,次级和第三纪蛋白结构的物理模型来计算结合亲和力,我们的模型从大量实验数据中学习了抗体序列和结合亲和力之间的映射。
然后,我们将模型的Pearson系数与先前作品的系数进行了比较16检查预测值与由于突变引起的实验亲和力变化之间的线性相关性(表1)。基于图的模型(Pearsonâ= 0.6)在计算机方法中的表现优于最常规的(基于结构),而基于语言的预测产生的Pearson系数为0.40,与Discovery Studio(Pearsonâ= 0.45)相当。这些发现表明,可以利用基于学习的深度表示来预测抗体成熟过程中完全未知变体的结合,这意味着该模型捕获了可转移的特征,从而有助于抗体 - 抗原相互作用的结合强度。
总而言之,基于图形和自然语言的方法都能够预测抗体成熟过程中与新型变体的相互作用。我们假设该模型捕获了可预测抗体 - 抗原相互作用中结合强度的关键转移特征。
亲和力成熟效率
我们提出的建模策略可以通过允许Sentinusal®以非常低的计算成本搜索大突变空间来大大降低复杂性。桌子2将硅亲和力成熟的效率与两种基于结构的方法的效率进行了比较。Prodigy(蛋白质结合能预测)是Web服务的集合,可预测生物复合物中结合亲和力的预测;他们还从晶体学信息中鉴定出生物界面31。同时,Schrodinger Desmond是一种用于计算自由能值的分子动力学工具(即结合亲和力)32。两种方法都需要复杂的结构输入。我们的方法使用任意抗体序列模板,该模板具有20个氨基酸的定义区域(ROI)。单位点突变是在ROI内进行的,然后进行所得突变空间的亲和力计算(筛选)。桌子2使用基于序列的方法和基于结构的方法,根据虚拟库的不同尺寸显示了计算时间成本。表2抗体成熟空间搜索的计算成本(序列长度
我们选择了具有最佳预测的RBD结合能力的前50个AI设计的抗体序列(AINL1-AINL50)。
这些序列在第一轮(前iickron)中合成并在功能上进行了评估。合成的大多数抗体能够与SARS-COV-2尖峰蛋白的RBD结合,通常在测试的最高浓度下达到过饱和态(即OD值> 2.0)。一些抗体也很好地与SARS-COV-2 RBD蛋白结合了较低的浓度\(0.03 \ mu \ text {g}/\ text {ml} \)(图 4)。第一个(AINL1-AINL50)和第二批(AINL51-AINL70)分别为50和20抗体,表达了良好的表达,并且与B.1,Delta和Omicron结合的命中率很高。第一批和第二批次产生\(14 \ text {\%} \)和\(40 \ text {\%} \)支持信息可在支持信息中提供三50抗(表S1)的第一轮OD450结果ELISA结果的ELISA结果(表S1)(表S2)。

OD 450的吸光度值来自50种抗体的直接ELISA(\(1 \ mu \ text {g}/\ text {ml} \);蓝色)反对野生类型(一个),三角洲(b)和Omicron(c)。盘子涂有\(1 \ text {ug}/\ text {ml} \)RBD蛋白。OD450\(= \)450 nm的光密度;two therapeutic SARS-CoV-2 antibodies were tested as controls (black, orange) for the variants.
We then measured the fold-change in binding affinity improvement of the designed antibody versus the template antibody as an indicator of affinity maturation performance (Fig. 5)。Qualitative differences in antibodies are reflected by the shape of dose-response curves for antibody binding (Fig. 6)。The original data for the dose responsive curve IC50 (Table S3) for the positive hits are also available in the supporting information.

Affinity changes in designed antibodies compared to their template antibodies (一个) Template 1 (CR3022) and associated AI-designed antibodies AINL53, AINL14, AINL54, and AINL 68. (b) Template 2 (Regen 10,933) and associated AI-designed antibodies AINL31, AINL38, AINL42, and AINL 40. (c) Template 3 (Regen 10,987) and associated AI-designed antibodies AINL45 and AINL48.图6

\(\text{and }1 \mu \text{g}/\text{ml}\)) of the RBD of WT, Delta, and Omicron.((一个) Controls of previously approved therapeutic antibodies;((b) first batch of AI-designed antibodies generated before Omicron prevalence;和 (c) second batch of AI-designed antibodies generated after Omicron prevalence.
Coronavirus cytopathic assay
We next measured the ability of the designed antibodies to reduce the of the Delta and Omicron strains infecting Vero E6 host cells (Fig. 7)。Ten antibodies neutralized the CPE of the Delta Strain at IC50 values of\(<10\text{ug}/\text{ml}\), and one antibody neutralized the CPE of Omicron (Fig. 7and Table3)。For example, the IC50 of AINNL0031 is\(2.704\text{ ug}/\text{mL}\), indicating that it strongly neutralizes the CPE.Overall, none of the antibodies were significantly directly toxic with > 10uM CC50 against Vero E6 host cells.The anti-viral assay and cytotoxicity assay results of the first round of 50 antibodies (Table S4) against Delta and all 70 antibodies (Table S5) against Omicron are available in supporting information.

CPE inhibition curves of selected antibodies against delta (一个) and Omicron (b)。The virus was neutralized by mixing a fixed number of infectious virus particles with serial dilutions of the antibody and then adding this mixture to Vera E6 cells.Luminescence was read using a Perkin Elmer envision or BMG CLARIOstar plate reader after a 10Â min incubation at room temperature to measure cell viability.
讨论
Here, we tested whether of an Al-based, structure-free approach can design antibodies that are effective in vitro and at drastically reduced time and cost compared to those designed via traditional antibody engineering and affinity maturation strategies.This structure-free approach is critical, because although a large number of proteins can be sequenced using current sequencing technologies, determining their crystal structures remains a complex, risky, and time-consuming task.Here, we employed an in-silico antibody discovery approach that uses affinity prediction models and deep learning techniques to design antibodies from complex sequence information alone (i.e., without any structural information).This modeling strategy captures functionally critical features encoded at the amino acid level that contribute to the interactions and the resulting binding affinities between antibody and antigen.Modeling at the amino acid rather than the atomic level significantly reduces the time needed for both training and prediction;moreover, it can also accommodate smaller annotated training datasets due to the model’s simplicity.
The computational workflow efficiency increase is because the trained model does not use any structural information from the protein sequences.Instead, only combinations of sequence strings are used as inputs for this model.Therefore, this allows the AI model to be trained on sequence text strings which are one-dimensional instead of a three-dimensional structure PDB file, simplifying the computational complexity of training this model.One item to note from this approach is that some of the training data used in this model may have some implicit bias when selecting the paratope from the entire protein sequence as the training set from SKEMPI or AB-Bind are manually labelled from known proteins.However, it is also noted that when running prediction tasks, labelled fragments of a protein are also used when known, such as when known extracellular or secreted part of a protein are labelled in UniProt.This further simplifies the computational efficiency, but also does not consider the explicit structure, but only considers the labelled results as a method for further simplification of our computational process.
The accuracy of our approach is higher than those of traditional methods;moreover, its reliability is also comparable to other methods featured in the benchmark study of traditional structure-based approaches on SKEMPI and AB-Bind datasets (Table1)。The results from the SARS-COV2 RBD work (Fig. 5) shows affinity improvements of 11.41-, 39.62-, and 269.65-fold compared to each original antibody template, even though the antigens were not part of the training set.These results demonstrate the model’s utility even for novel antigens.In addition, the resulting scores of the computation process is compared to the actual ELISA OD450 assay value shown in Fig. 8。In this figure, scatterplots are drawn for both Delta and Omicron antibodies.It can be seen that the top scoring AI antibodies are also positively correlated to the actual ELISA value, especially when comparing some of the most active antibodies confirmed using this assay (OD450 > 4).Although false positives are also found, this shows a much-improved overall hit rate compared to traditional methods such as using hybridoma clones.In a study for screening and selecting hybridomas producing antibodies targeting PD-1, only 51 out of 10,560 pools are identified as positive hits for their ELISA assay33。In terms of a secondary screening using flow cytometry, this workflow produced 5 hits from the primary 51 hits33。When directly comparing to our AI-based method, material costs and experimentation time with cell culture and hybridoma generation are directly saved as the AI workflow directly outputs monoclonal antibodies for expression and binding assays and resulting in an overall higher hit rate with significantly less samples for experimentation.

ELISA assay OD450 values compared to the model’s predicted score.Both delta and Omicron data are displayed in the scatterplots.The model has successfully predicted many of the ELISA-validated active antibodies (ELISA OD450 > 4) as the top-ranking model prediction scores (model score > 0.6).
Interestingly, we found a discrepancy between the antibody binding data obtained through ELISA and the observed neutralizing capacity of the antibody for blocking infection.This difference may be due to a number of factors, including differences in RBD structure when the protein is plate-bound in the ELISA versus its structure when interacting with a live virus or in the presence of different antibody binding sites on the RBD (e.g., antibodies having high-affinity interactions that may not physically block the RBD and receptor interaction).
Our Al model was also able to design cross-binding antibodies against many different antigen populations, including viral mutant strains.The cross-binding antibodies may be capable of mutation-resistant binding to future evolving RBD region variations;therefore, the neutralization potency of such antibodies may be broad, which is a key characteristic of therapeutic antibodies targeting rapidly mutating viruses such as SARS-CoV-2.We hypothesized that the high dimensional features learned during the Al training process may represent components of the viral evolutionary process.This results in an ability to predict binding affinity in the virtual screening process even though the process considers only known virus strains.This approach was successful for SARS-COV-2, where\(14\text{\%}\)of the 50 screened antibodies generated prior to Omicron were able to bind to all the strains (Omicron, Delta, and wild-type).Cross-reacting antibodies may also be less specific34;however, a broadly cross-reactive antibody (e.g., generated through engineering approaches35) may have better therapeutic potential in coping with viral evolution.
Our computational workflow allows for iterations with wet laboratory results to achieve better antibody design and cross-validation.This was demonstrated by the 40% cross-binding hit rate obtained in the second round of antibody synthesis (of 20 sequences), a considerable performance leap compared to the 14% cross-binding hit rate in the first round.
We have demonstrated a highly efficient and cost-effective approach for generating therapeutic antibodies for single or multiple viral strains.However, during the evaluation of this approach’s effectiveness, we found that the neutralizing capability of the designed antibody is not consistent with the binding affinity results.Aside from binding affinity, there are multiple mechanisms and processes involved in the neutralizing effects.For example, the binding epitope on the spike protein and antibody conformation also impact the infection and translocation process during viral invasion.Therefore, epitope mapping and conformation dynamics studies are required for a more precise design of neutralizing antibodies.In addition, we have not performed in vivo efficacy studies, which was beyond the scope of this research.
This study presents a novel approach with abilities to model the initial design of therapeutic antibodies and to iteratively improve upon these initial designs to account for future mutations in the target protein of a rapidly evolving pathogenic virus.Because our approach combines flexibility and high-throughput at a low computational cost, it can be beneficial in other applications of the technology as well.
数据可用性
All data generated or analyzed during this study are included in this published article and its supplementary information files.
参考
Deng, X. et al.Transmission, infectivity, and antibody neutralization of an emerging SARS-COV-2 variant in California carrying a L452r spike protein mutation.细胞 184, 3426-3437.e8 (2021).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Quiros-Roldan, E. et al.Monoclonal antibodies against SARS-CoV-2: current scenario and future perspectives.药品 14, 1272 (2021).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Choi, J. Y. & Smith, D. M. SARS-CoV-2 variants of concern.Yonsei Med.J. 62, 961 (2021).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Wu, Z. et al.A comprehensive survey on graph neural networks.IEEE Trans。Neural Netw.学习。系统。 32, 4–24 (2020).
文章一个 MathSciNet一个 Google Scholar一个
Fan, W. et al.Graph Neural Networks for Social Recommendation(ACM, 2019).
书一个 Google Scholar一个
Gao, H., Chen, Y. & Ji, S.Learning Graph Pooling and Hybrid Convolutional Operations for Text Representations(ACM, 2019).
书一个 Google Scholar一个
Zhang, Z. et al.Relational graph neural network with hierarchical attention for knowledge graph completion.Proc。int。aaai conf。 34, 9612–9619 (2020).
Desmond, J. H., RaÅ¡ajski, M. & Pržulj, N. Fitting a geometric graph to a proteinprotein interaction network.生物信息学 24, 1093–1099 (2008).
文章一个 Google Scholar一个
Liu, J. & Gong, X. Attention mechanism enhanced LSTM with residual architecture and its application for protein-protein interaction residue pairs prediction.BMC Bioinform. 20, 111 (2019).
文章一个 Google Scholar一个
Saka, K. Antibody design using LSTM based deep generative model from phage display library for affinity maturation.科学。代表。 11, 1–13 (2021).
文章一个 广告一个 Google Scholar一个
Noumi, T. Epitope prediction of antigen protein using attention-based LSTM network.J. Info.Proc。 29, 321–327 (2021).
Ofer, D., Brandes, N. & Linial, M. The language of proteins: NLP, machine learning & protein sequences.计算。结构。生物技术。J. 19, 1750–1758 (2021).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Ahmed, E., et al.ProtTrans: towards cracking the language of lifes code through self-supervised deep learning and high performance computing.(2020)。
Yadav, S., Ekbal, A., Saha, S., Kumar, A. & Bhattacharyya, P. Feature assisted stacked attentive shortest dependency path based Bi-LSTM model for protein-protein interaction.知识。Based Syst. 166, 18–29 (2019).
文章一个 Google Scholar一个
JankauskaitÄ—, J., Jiménez-GarcÃa, B., DapkÅ«nas, J., Fernández-Recio, J. & Moal, I. H. SKEMPI 2.0: an updated benchmark of changes in protein-protein binding energy, kinetics and thermodynamics upon mutation.生物信息学 35, 462–469 (2019).
文章一个 PubMed一个 Google Scholar一个
Sirin, S., Apgar, J. R., Bennett, E. M. & Keating, A. E. AB -bind: antibody binding mutational database for computational affinity predictions.蛋白质科学。 25, 393–409 (2016).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Olsen, T. H., Boyles, F. & Deane, C. M. Observed antibody space: a diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences.蛋白质科学。 31, 141–146 (2022).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Wang, Y. et al.A crowdsourcing open platform for literature curation in UniProt.Plos Biol。 19, e3001464 (2021).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Leng, D., Guo, J., Pan, L., Li, J. & Wang, X. Enhance information propagation for graph neural network by heterogeneous aggregations.arxiv1–8 (2021).
Elnaggar, A. et al.ProtTrans: Toward understanding the language of life through self-supervised learning.IEEE Trans。模式肛门。Mach.Intell。 44(10), 7112–7127 (2021).
文章一个 Google Scholar一个
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B. & Wu, C. H. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches.Bioinformatic 31(6), 926–932 (2015).
文章一个 CAS一个 Google Scholar一个
Khare, S. et al.GISAID’s role in pandemic response.中国CDC周刊 3, 1049 (2021).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Elbe, S. & Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health.地球。Chall. 1, 33–46 (2017).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Shu, Y. & Mccauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality.Eurosurveillance 22, 30494 (2017).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Ter Meulen, J. et al.Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants.Plos Med。 3, e237 (2006).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Jiangdong, H., Owens, R. & Stuart, J. N.Single Domain Antibodies Binding to Sars-Cov-2 Spike Protein。United States Patent Application US 17/923,142 (1990).
Ter Meulen, J. et al.Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets.柳叶刀 363, 2139–2141 (2004).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Ganguly, S. et al.Regeneron pharmaceuticals.在Methods for Treating or Preventing Sars-Cov-2 Infections and Covid-19 with Anti-Sars-Cov-2 Spike Glycoprotein Antibodies(eds Ganguly, S. et al.) (Google Patents, 2021).
Deeks, E. D. Casirivimab/Imdevimab: first approval.毒品 81, 2047–2055 (2021).
文章一个 CAS一个 PubMed一个 PubMed Central一个 Google Scholar一个
Dunbar, J. & Deane, C. M. ANARCI: antigen receptor numbering and receptor classification.生物信息学 32(2), 298–300 (2016).
文章一个 CAS一个 PubMed一个 Google Scholar一个
Honorato, R. V. et al.Structural biology in the clouds: the WeNMR-EOSC ecosystem.正面。摩尔。Biosci。 8, 729513 (2021).
文章一个 PubMed一个 PubMed Central一个 Google Scholar一个
Schrödinger Release 2022–3: Desmond Molecular Dynamics System, D. E. Shaw Research.Maestro-Desmond Interoperab.工具(2021)。
Phakham, T. et al.Highly efficient hybridoma generation and screening strategy for anti-PD-1 monoclonal antibody development.纳特。科学。代表。 12, 17792 (2022).
广告一个 CAS一个 Google Scholar一个
Buchwalow, I., Samoilova, V., Boecker, W. & Tiemann, M. Non-specific binding of antibodies in immunohistochemistry: fallacies and facts.科学。代表。 1, 1–6 (2011).
文章一个 Google Scholar一个
Leivo, J., Chappuis, C., Lamminmäki, U., Lövgren, T. & Vehniäinen, M. Engineering of a broad-specificity antibody: detection of eight fluoroquinolone antibiotics simultaneously.肛门。生物化学。 409, 14–21 (2011).
文章一个 CAS一个 PubMed一个 Google Scholar一个
致谢
We thank Junfeng Wu, Pinyu Xiao, Roger Wang, Xinyu Wang and Vincent Brand for their engineering effort on the SentinusAl® platform.We thank Dr. Sheng Ding, Dr. Iwan Alexander, Dr. Murat Tanik, Dr. Yang Jiao, Dr. Limeng Pu and Dr. Paige Vinson for their scientific support on Ainnocence’s research works.The abovementioned 70 antibody sequences are filed in the application for Patent Cooperation Treaty (PCT) (Application id: PCT/CN2022/094,029).The abovementioned AI protein design system is filed in US Patents: US20230377689A1, US US20240047006A1.
道德声明
竞争利益
作者没有宣称没有竞争利益。
附加信息
Publisher’s note
关于已发表的地图和机构隶属关系中的管辖权主张,Springer自然仍然是中立的。
补充信息
权利和权限
开放访问This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.要查看此许可证的副本,请访问http://creativecommons.org/licenses/by/4.0/。重印和权限
引用本文

Kang, Y., Jin, K. & Pan, L. AI designed, mutation resistant broad neutralizing antibodies against multiple SARS-CoV-2 strains.
Sci代表15 , 15533 (2025).https://doi.org/10.1038/s41598-025-98979-w
已收到:
公认:
出版:
doi:https://doi.org/10.1038/s41598-025-98979-w