
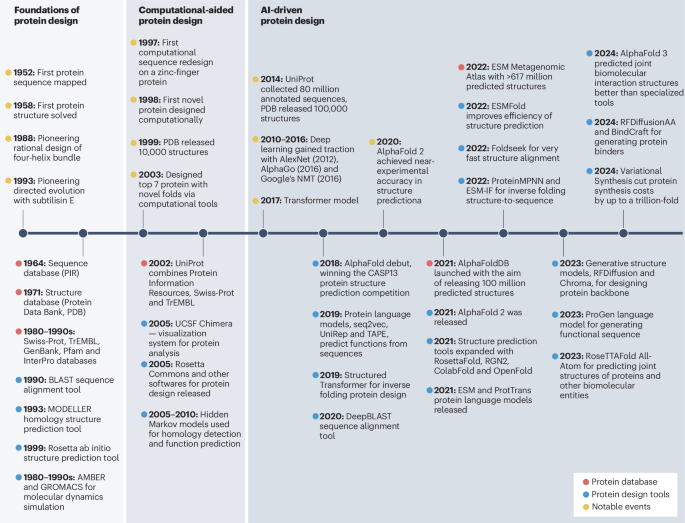
AI-driven protein design
作者:Church, George M.
References
Ebrahimi, S. B. & Samanta, D. Engineering protein-based therapeutics through structural and chemical design. Nat. Commun. 14, 2411 (2023).
Chen, K. & Arnold, F. H. Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc. Natl Acad. Sci. USA 90, 5618–5622 (1993).
Lajoie, M. J. et al. Genomically recoded organisms expand biological functions. Science 342, 357–360 (2013).
Listov, D., Goverde, C. A., Correia, B. E. & Fleishman, S. J. Opportunities and challenges in design and optimization of protein function. Nat. Rev. Mol. Cell Biol. 25, 639–653 (2024).
Alley, E. C., Khimulya, G., Biswas, S., AlQuraishi, M. & Church, G. M. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 16, 1315–1322 (2019). UniRep is one of the first protein language models to learn rich evolutionary, structural and biophysical representations from raw, unlabelled protein sequences, demonstrating how such models can power a diverse suite of artificial intelligence-driven tools.
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). AlphaFold 2 is the first model to regularly predict protein 3D structures from amino-acid sequences with near-experimental accuracy, and its high-fidelity structural predictions now underpin artificial intelligence-driven protein design workflows.
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Dauparas, J. et al. Robust deep learning-based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022). ProteinMPNN solves the inverse folding challenge by generating amino-acid sequences for fixed backbones with accuracy well above physics-based methods and at high throughput, making it a widely adopted cornerstone in artificial intelligence-driven rational design workflows.
Watson, J. L. et al. De novo design of protein structure and function with RFDiffusion. Nature 620, 1089–1100 (2023). RFDiffusion generates protein backbones that meet specified structural or functional objectives with high success rates across diverse, experimentally validated design settings, including de novo design.
Hamamsy, T. et al. Protein remote homology detection and structural alignment using deep learning. Nat. Biotechnol. 42, 975–985 (2024).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2024).
Wayment-Steele, H. K. et al. Predicting multiple conformations via sequence clustering and AlphaFold2. Nature 625, 832–839 (2024).
Krishna, R. et al. Generalized biomolecular modeling and design with RoseTTAFold All-Atom. Science 384, eadl2528 (2024).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Hutchison, C. A. et al. Mutagenesis at a specific position in a DNA sequence. J. Biol. Chem. 253, 6551–6560 (1978).
Alber, T., Sun, D. P., Nye, J. A., Muchmore, D. C. & Matthews, B. W. Temperature-sensitive mutations of bacteriophage T4 lysozyme occur at sites with low mobility and low solvent accessibility in the folded protein. Biochemistry 26, 3754–3758 (1987).
Marshall, S. A., Lazar, G. A., Chirino, A. J. & Desjarlais, J. R. Rational design and engineering of therapeutic proteins. Drug Discov. Today 8, 212–221 (2003).
Davey, J. A., Damry, A. M., Goto, N. K. & Chica, R. A. Rational design of proteins that exchange on functional timescales. Nat. Chem. Biol. 13, 1280–1285 (2017).
Yang, K. K., Wu, Z. & Arnold, F. H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694 (2019).
Huang, P.-S., Boyken, S. E. & Baker, D. The coming of age of de novo protein design. Nature 537, 320–327 (2016).
Hie, B. L. et al. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 42, 275–283 (2024).
Koh, H. Y., Nguyen, A. T. N., Pan, S., May, L. T. & Webb, G. I. Physicochemical graph neural network for learning protein–ligand interaction fingerprints from sequence data. Nat. Mach. Intell. 6, 673–687 (2024).
Chowdhury, R. et al. Single-sequence protein structure prediction using a language model and deep learning. Nat. Biotechnol. 40, 1617–1623 (2022).
Ingraham, J. B. et al. Illuminating protein space with a programmable generative model. Nature 623, 1070–1078 (2023).
Chai Discovery Team et al. Chai-1: decoding the molecular interactions of life. Preprint at bioRxiv https://doi.org/10.1101/2024.10.10.615955 (2024).
Bryant, D. H. et al. Deep diversification of an AAV capsid protein by machine learning. Nat. Biotechnol. 39, 691–696 (2021). This study applies AI-driven directed evolution to generate and screen ~1010 AAV2 capsid variants, yielding 110,689 viable mutants that exceed natural serotype diversity, and positions AI-driven capsid diversification as a new paradigm in gene-therapy vector engineering.
Ogden, P. J., Kelsic, E. D., Sinai, S. & Church, G. M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 366, 1139–1143 (2019).
Jiang, K. et al. Rapid in silico directed evolution by a protein language model with EVOLVEpro. Science 387, eadr6006 (2024). This study optimizes artificial intelligence-driven directed evolution by integrating protein language-model embeddings with sequence-based activity predictors, achieving up to 100-fold improvements in protein activity across diverse targets and streamlining modern directed evolution workflows.
Yang, J. et al. Active learning-assisted directed evolution. Nat. Commun. 16, 714 (2025).
Gainza, P. et al. De novo design of protein interactions with learned surface fingerprints. Nature 617, 176–184 (2023). This study developed a unified artificial intelligence-driven rational design workflow that integrates 3D geometric network for binding-site prediction, structural database mining and motif-based binder design to generate de novo protein binders against targets such as the SARS-CoV-2 spike with nanomolar affinities.
Grøn, H., Bech, L. M., Branner, S. & Breddam, K. A highly active and oxidation-resistant subtilisin-like enzyme produced by a combination of site-directed mutagenesis and chemical modification. Eur. J. Biochem. 194, 897–901 (1990).
Fleishman, S. J. et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332, 816–821 (2011).
Varadi, M. et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023). This study introduces ESM2, one of the most widely adopted protein language models, and ESMFold, which matches AlphaFold 2’s accuracy using only single‐sequence inputs without multiple‐sequence alignments, enabling substantially faster structure prediction.
Hayes, T. et al. Simulating 500 million years of evolution with a language model. Science 387, 850–858 (2025).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Ravindra et al. Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nat. Methods 17, 541–550 (2020).
Silva, D.-A. et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature 565, 186–191 (2019).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Kaminski, K., Ludwiczak, J., Pawlicki, K., Alva, V. & Dunin-Horkawicz, S. pLM-BLAST: distant homology detection based on direct comparison of sequence representations from protein language models. Bioinformatics 39, btad579 (2023).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Holm, L. Dali server: structural unification of protein families. Nucleic Acids Res. 50, W210–W215 (2022).
The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019).
Hopf, T. A. et al. The EVcouplings Python framework for coevolutionary sequence analysis. Bioinformatics 35, 1582–1584 (2019).
Burley, S. K. et al. RCSB Protein Data Bank (RCSB.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 51, D488–D508 (2023).
Baek, M. et al. Accurate prediction of protein–nucleic acid complexes using RoseTTAFoldNA. Nat. Methods 21, 117–121 (2024).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. Preprint at bioRxiv https://doi.org/10.1101/2021.10.04.463034 (2022).
Radivojac, P. et al. A large-scale evaluation of computational protein function prediction. Nat. Methods 10, 221–227 (2013).
Gainza, P. et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 17, 184–192 (2020).
Weinstein, E. N. et al. Manufacturing-aware generative model architectures enable biological sequence design and synthesis at petascale. Preprint at bioRxiv https://doi.org/10.1101/2024.09.13.612900 (2024).
Packer, M. S. & Liu, D. R. Methods for the directed evolution of proteins. Nat. Rev. Genet. 16, 379–394 (2015).
Biswas, S., Khimulya, G., Alley, E. C., Esvelt, K. M. & Church, G. M. Low-N protein engineering with data-efficient deep learning. Nat. Methods 18, 389–396 (2021).
Madani, A. et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 41, 1099–1106 (2023). ProGen shows that large protein language models conditioned on ‘tags’ (short textual annotations such as enzyme function) can generate functional protein sequences across diverse families, enabling rapid tag-driven protein design without explicit structural input.
Yeh, A. H.-W. et al. De novo design of luciferases using deep learning. Nature 614, 774–780 (2023). This study integrates AI tools such as structure prediction, sequence design and virtual screening into a unified AI-driven rational design workflow to create de novo luciferases that catalyse DTZ chemiluminescence with exceptional specificity.
Cao, L. et al. Design of protein-binding proteins from the target structure alone. Nature 605, 551–560 (2022).
Shanker, V. R., Bruun, T. U. J., Hie, B. L. & Kim, P. S. Unsupervised evolution of protein and antibody complexes with a structure-informed language model. Science 385, 46–53 (2024).
Röthlisberger, D. et al. Kemp elimination catalysts by computational enzyme design. Nature 453, 190–195 (2008).
Lauko, A. et al. Computational design of serine hydrolases. Science 388, eadu2454 (2025).
Smith, T. F. & Waterman, M. S. Identification of common molecular subsequences. J. Mol. Biol. 147, 195–197 (1981).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Llinares-López, F., Berthet, Q., Blondel, M., Teboul, O. & Vert, J.-P. Deep embedding and alignment of protein sequences. Nat. Methods 20, 104–111 (2023).
Liu, W. et al. PLMSearch: protein language model powers accurate and fast sequence search for remote homology. Nat. Commun. 15, 2775 (2024).
Kim, W. et al. Rapid and sensitive protein complex alignment with Foldseek-Multimer. Nat. Methods 22, 469–472 (2025).
van den Oord, A., Vinyals, O. & kavukcuoglu, K. Neural discrete representation learning. In Advances in Neural Information Processing Systems (eds Guyon, I. et a.) Vol. 30 (Curran Associates, 2017).
Eom, H. et al. Discovery of highly active kynureninases for cancer immunotherapy through protein language model. Nucleic Acids Res. 53, gkae1245 (2025).
Hu, M. et al. Advances in Neural Information Processing Systems Vol. 35 (Curran Associates, Inc., 2022).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Ahdritz, G. et al. OpenFold: retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. Nat. Methods 21, 1514–1524 (2024).
Ketata, M. A. et al. DiffDock-PP: rigid protein–protein docking with diffusion models. Preprint at https://doi.org/10.48550/arXiv.2304.03889 (2023).
Qiao, Z., Nie, W., Vahdat, A., Miller, T. F. & Anandkumar, A. State-specific protein–ligand complex structure prediction with a multiscale deep generative model. Nat. Mach. Intell. 6, 195–208 (2024).
Guo, H.-B. et al. AlphaFold2 models indicate that protein sequence determines both structure and dynamics. Sci. Rep. 12, 10696 (2022).
Anishchenko, I. et al. De novo protein design by deep network hallucination. Nature 600, 547–552 (2021).
Wang, J. et al. Scaffolding protein functional sites using deep learning. Science 377, 387–394 (2022).
He, J., Turzo, S. B. A., Seffernick, J. T., Kim, S. S. & Lindert, S. Prediction of intrinsic disorder using Rosetta ResidueDisorder and AlphaFold2. J. Phys. Chem. B 126, 8439–8446 (2022).
Kurgan, L. et al. Tutorial: a guide for the selection of fast and accurate computational tools for the prediction of intrinsic disorder in proteins. Nat. Protoc. 18, 3157–3172 (2023).
Vander Meersche, Y., Cretin, G., de Brevern, A. G., Gelly, J.-C. & Galochkina, T. MEDUSA: prediction of protein flexibility from sequence. J. Mol. Biol. 433, 166882 (2021).
Mészáros, B., Erdős, G. & Dosztányi, Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46, W329–W337 (2018).
Hu, G. et al. flDPnn: accurate intrinsic disorder prediction with putative propensities of disorder functions. Nat. Commun. 12, 4438 (2021).
Roney, J. P. & Ovchinnikov, S. State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101 (2022).
Pak, M. A. et al. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS ONE 18, e0282689 (2023).
Pudžiuvelytė, I. et al. TemStaPro: protein thermostability prediction using sequence representations from protein language models. Bioinformatics 40, btae157 (2024).
Blaabjerg, L. M. et al. Rapid protein stability prediction using deep learning representations. eLife 12, e82593 (2023).
Zhou, Y., Pan, Q., Pires, D. E. V., Rodrigues, C. H. M. & Ascher, D. B. DDMut: predicting effects of mutations on protein stability using deep learning. Nucleic Acids Res. 51, W122–W128 (2023).
Yin, R., Feng, B. Y., Varshney, A. & Pierce, B. G. Benchmarking AlphaFold for protein complex modeling reveals accuracy determinants. Protein Sci. 31, e4379 (2022).
Ferreiro, D. U., Komives, E. A. & Wolynes, P. G. Frustration in biomolecules. Q. Rev. Biophys. 47, 285–363 (2014).
del Alamo, D., Sala, D., Mchaourab, H. S. & Meiler, J. Sampling alternative conformational states of transporters and receptors with AlphaFold2. eLife 11, e75751 (2022).
Guan, X. et al. Predicting protein conformational motions using energetic frustration analysis and AlphaFold2. Proc. Natl Acad. Sci. USA 121, e2410662121 (2024).
Chakravarty, D. et al. AlphaFold predictions of fold-switched conformations are driven by structure memorization. Nat. Commun. 15, 7296 (2024).
Jing, B., Berger, B. & Jaakkola, T. AlphaFold meets flow matching for generating protein ensembles. In Proc. 41st International Conference on Machine Learning Vol. 235, 22277–22303 (JMLR.org, 2024).
Wang, T. et al. Ab initio characterization of protein molecular dynamics with AI2BMD. Nature 635, 1019–1027 (2024).
Wang, Y. et al. Enhancing geometric representations for molecules with equivariant vector–scalar interactive message passing. Nat. Commun. 15, 313 (2024).
Arnold, C. AlphaFold touted as next big thing for drug discovery — but is it? Nature 622, 15–17 (2023).
Callaway, E. Major AlphaFold upgrade offers boost for drug discovery. Nature 629, 509–510 (2024).
Miller, E. B. et al. Enabling structure-based drug discovery utilizing predicted models. Cell 187, 521–525 (2024).
Jang, Y. J. et al. Accurate prediction of protein function using statistics-informed graph networks. Nat. Commun. 15, 6601 (2024).
You, R. et al. NetGO: improving large-scale protein function prediction with massive network information. Nucleic Acids Res. 47, W379–W387 (2019).
Yao, S. et al. NetGO 2.0: improving large-scale protein function prediction with massive sequence, text, domain, family and network information. Nucleic Acids Res. 49, W469–W475 (2021).
Wang, S., You, R., Liu, Y., Xiong, Y. & Zhu, S. NetGO 3.0: protein language model improves large-scale functional annotations. Genom. Proteom. Bioinform. 21, 349–358 (2023).
Le Guilloux, V., Schmidtke, P. & Tuffery, P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinform. 10, 168 (2009).
Porollo, A. & Meller, J. Prediction-based fingerprints of protein–protein interactions. Proteins Struct. Funct. Bioinform. 66, 630–645 (2007).
Murakami, Y. & Mizuguchi, K. Applying the naive Bayes classifier with kernel density estimation to the prediction of protein–protein interaction sites. Bioinformatics 26, 1841–1848 (2010).
Tubiana, J., Schneidman-Duhovny, D. & Wolfson, H. J. ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction. Nat. Methods 19, 730–739 (2022).
Jiménez, J., Doerr, S., Martínez-Rosell, G., Rose, A. S. & De Fabritiis, G. DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 33, 3036–3042 (2017).
Corso, G., Stärk, H., Jing, B., Barzilay, R. & Jaakkola, T. DiffDock: diffusion steps, twists, and turns for molecular docking. In International Conference on Learning Representations (2023).
Elliott, S. et al. Enhancement of therapeutic protein in vivo activities through glycoengineering. Nat. Biotechnol. 21, 414–421 (2003).
Hunter, T. The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 (2007).
Ramazi, S. & Zahiri, J. Post-translational modifications in proteins: resources, tools and prediction methods. Database 2021, baab012 (2021).
Wang, D. et al. MusiteDeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 33, 3909–3916 (2017).
Wang, D. et al. MusiteDeep: a deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 48, W140–W146 (2020).
Shrestha, P., Kandel, J., Tayara, H. & Chong, K. T. Post-translational modification prediction via prompt-based fine-tuning of a GPT-2 model. Nat. Commun. 15, 6699 (2024).
Yan, Y. et al. MIND-S is a deep-learning prediction model for elucidating protein post-translational modifications in human diseases. Cell Rep. Methods 3, 100430 (2023).
Shi, X.-X. et al. PTMdyna: exploring the influence of post-translation modifications on protein conformational dynamics. Brief. Bioinform. 23, bbab424 (2022).
Zhou, N. et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 20, 244 (2019).
Bloom, J. D., Labthavikul, S. T., Otey, C. R. & Arnold, F. H. Protein stability promotes evolvability. Proc. Natl Acad. Sci. USA 103, 5869–5874 (2006).
Meier, J. et al. Advances in Neural Information Processing Systems Vol. 34, 29287–29303 (Curran Associates, Inc., 2021).
Ferruz, N., Schmidt, S. & Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 13, 4348 (2022).
Unsal, S. et al. Learning functional properties of proteins with language models. Nat. Mach. Intell. 4, 227–245 (2022).
Ferruz, N. & Höcker, B. Controllable protein design with language models. Nat. Mach. Intell. 4, 521–532 (2022).
Truong, T. F. Jr & Bepler, T. PoET: A generative model of protein families as sequences-of-sequences. In Advances in Neural Information Processing Systems (eds Oh, A. et al.) Vol. 36 (Curran Associates, 2023).
Gligorijević, V. et al. Function-guided protein design by deep manifold sampling. Preprint at bioRxiv https://doi.org/10.1101/2021.12.22.473759 (2021).
Kucera, T., Togninalli, M. & Meng-Papaxanthos, L. Conditional generative modeling for de novo protein design with hierarchical functions. Bioinformatics 38, 3454–3461 (2022).
Ingraham, J., Garg, V., Barzilay, R. & Jaakkola, T. Generative models for graph-based protein design. In Advances in Neural Information Processing Systems (eds Wallach, H. et al.) Vol. 32 (Curran Associates, 2019).
Hsu, C. et al. Learning inverse folding from millions of predicted structures. In Proc. 39th International Conference on Machine Learning 8946–8970 (PMLR, 2022).
Dauparas, J. et al. Atomic context-conditioned protein sequence design using LigandMPNN. Nat. Methods 22, 717–723 (2025).
McFerrin, L. & Ratan, U. Highlights from the AWS Life Sciences Executive Symposium 2023: accelerating pharma drug discovery with ML and generative AI. AWS Blogs https://go.nature.com/4gbiXvp (31 May 2023).
Goverde, C. A. et al. Computational design of soluble and functional membrane protein analogues. Nature 631, 449–458 (2024).
Dou, J. et al. De novo design of a fluorescence-activating β-barrel. Nature 561, 485–491 (2018).
Gao, B. et al. Advances in Neural Information Processing Systems Vol. 36 (Curran Associates, Inc., 2023).
Ho, J., Jain, A. & Abbeel, P. Advances in Neural Information Processing Systems Vol. 33 (Curran Associates, Inc., 2020).
Trippe, B. L. et al. Diffusion probabilistic modeling of protein backbones in 3D for the motif-scaffolding problem. Int. Conf. Learn. Represent. ICLR 2022 (2022).
Luo, S. et al. Antigen-specific antibody design and optimization with diffusion-based generative models for protein structures. Adv. Neural Inf. Process. Syst. 35, 9754–9767 (2022).
Yang, J. et al. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl Acad. Sci. USA 117, 1496–1503 (2020).
Bennett, N. R. et al. Improving de novo protein binder design with deep learning. Nat. Commun. 14, 2625 (2023).
Pacesa, M. et al. BindCraft: one-shot design of functional protein binders. Preprint at bioRxiv https://doi.org/10.1101/2024.09.30.615802 (2024).
Wicky, B. I. M. et al. Hallucinating symmetric protein assemblies. Science 378, 56–61 (2022).
Lisanza, S. L. et al. Multistate and functional protein design using RoseTTAFold sequence space diffusion. Nat. Biotechnol. 43, 1288–1298 (2024).
Chu, A. E. et al. An all-atom protein generative model. Proc. Natl Acad. Sci. USA 121, e2311500121 (2024).
McNutt, A. T. et al. GNINA 1.0: molecular docking with deep learning. J. Cheminform. 13, 43 (2021).
Zhou, Z. et al. Enhancing efficiency of protein language models with minimal wet-lab data through few-shot learning. Nat. Commun. 15, 5566 (2024).
Hsu, C., Nisonoff, H., Fannjiang, C. & Listgarten, J. Learning protein fitness models from evolutionary and assay-labeled data. Nat. Biotechnol. 40, 1114–1122 (2022).
Frey, N. C. et al. Lab-in-the-loop therapeutic antibody design with deep learning. Preprint at bioRxiv https://doi.org/10.1101/2025.02.19.639050 (2025).
Wu, Z., Kan, S. B. J., Lewis, R. D., Wittmann, B. J. & Arnold, F. H. Machine learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl Acad. Sci. USA 116, 8852–8858 (2019).
Narayanan, H. et al. Machine learning for biologics: opportunities for protein engineering, developability, and formulation. Trends Pharmacol. Sci. 42, 151–165 (2021).
Gentiluomo, L. et al. Application of interpretable artificial neural networks to early monoclonal antibodies development. Eur. J. Pharm. Biopharm. 141, 81–89 (2019).
Gentiluomo, L., Roessner, D. & Frieß, W. Application of machine learning to predict monomer retention of therapeutic proteins after long term storage. Int. J. Pharm. 577, 119039 (2020).
Wang, C. & Zou, Q. Prediction of protein solubility based on sequence physicochemical patterns and distributed representation information with DeepSoluE. BMC Biol. 21, 12 (2023).
Zhang, X. et al. PLM_Sol: predicting protein solubility by benchmarking multiple protein language models with the updated Escherichia coli protein solubility dataset. Brief. Bioinform. 25, bbae404 (2024).
Planas-Iglesias, J. et al. AggreProt: a web server for predicting and engineering aggregation prone regions in proteins. Nucleic Acids Res. 52, W159–W169 (2024).
Louros, N., Schymkowitz, J. & Rousseau, F. Mechanisms and pathology of protein misfolding and aggregation. Nat. Rev. Mol. Cell Biol. 24, 912–933 (2023).
Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454 (2020).
Hashemi, N. et al. Improved prediction of MHC-peptide binding using protein language models. Front. Bioinform. 3, 1207380 (2023).
Müller, M. et al. Machine learning methods and harmonized datasets improve immunogenic neoantigen prediction. Immunity 56, 2650–2663.e6 (2023).
Li, G., Iyer, B., Prasath, V. B. S., Ni, Y. & Salomonis, N. DeepImmuno: deep learning-empowered prediction and generation of immunogenic peptides for T-cell immunity. Brief. Bioinform. 22, bbab160 (2021).
Marks, C., Hummer, A. M., Chin, M. & Deane, C. M. Humanization of antibodies using a machine learning approach on large-scale repertoire data. Bioinformatics 37, 4041–4047 (2021).
Qiu, Y. & Cheng, F. Artificial intelligence for drug discovery and development in Alzheimer’s disease. Curr. Opin. Struct. Biol. 85, 102776 (2024).
Zambaldi, V. et al. De novo design of high-affinity protein binders with AlphaProteo. Preprint at https://doi.org/10.48550/arXiv.2409.08022 (2024).
Ostrov, N. et al. Design, synthesis, and testing toward a 57-codon genome. Science 353, 819–822 (2016).
Liu, Y., Yang, Q. & Zhao, F. Synonymous but not silent: the codon usage code for gene expression and protein folding. Annu. Rev. Biochem. 90, 375–401 (2021).
Hanson, G. & Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 19, 20–30 (2018).
Fu, H. et al. Codon optimization with deep learning to enhance protein expression. Sci. Rep. 10, 17617 (2020).
Sidi, T., Bahiri-Elitzur, S., Tuller, T. & Kolodny, R. Predicting gene sequences with AI to study codon usage patterns. Proc. Natl Acad. Sci. USA 122, e2410003121 (2025).
Constant, D. A. et al. Deep learning-based codon optimization with large-scale synonymous variant datasets enables generalized tunable protein expression. Preprint at bioRxiv https://doi.org/10.1101/2023.02.11.528149 (2023).
Ren, Z. et al. CodonBERT: a BERT-based architecture tailored for codon optimization using the cross-attention mechanism. Bioinformatics 40, btae330 (2024).
Fallahpour, A., Gureghian, V., Filion, G. J., Lindner, A. B. & Pandi, A. CodonTransformer: a multispecies codon optimizer using context-aware neural networks. Nat. Commun. 16, 3205 (2025).
Weinstein, E. N. et al. Optimal design of stochastic DNA synthesis protocols based on generative sequence models. In Proc. 25th International Conference on Artificial Intelligence and Statistics 7450–7482 (PMLR, 2022).
Stark, H., Padia, U., Balla, J., Diao, C. & Church, G. CodonMPNN for organism specific and codon optimal inverse folding. Preprint at https://doi.org/10.48550/arXiv.2409.17265 (2024).
Outeiral, C. & Deane, C. M. Codon language embeddings provide strong signals for use in protein engineering. Nat. Mach. Intell. 6, 170–179 (2024).
Nguyen, E. et al. Sequence modeling and design from molecular to genome scale with Evo. Science 386, eado9336 (2024).
Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860 (2017).
Mendell, J. R. et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 377, 1713–1722 (2017).
Ding, F. & Steinhardt, J. Protein language models are biased by unequal sequence sampling across the tree of life. Preprint at bioRxiv https://doi.org/10.1101/2024.03.07.584001 (2024).
Volkov, M. et al. On the frustration to predict binding affinities from protein–ligand structures with deep neural networks. J. Med. Chem. 65, 7946–7958 (2022).
Medina-Ortiz, D., Khalifeh, A., Anvari-Kazemabad, H. & Davari, M. D. Interpretable and explainable predictive machine learning models for data-driven protein engineering. Biotechnol. Adv. 79, 108495 (2025).
Simon, E. & Zou, J. InterPLM: discovering interpretable features in protein language models via sparse autoencoders. Preprint at bioRxiv https://doi.org/10.1101/2024.11.14.623630 (2025).
AI’s potential to accelerate drug discovery needs a reality check. Nature 622, 217–217 (2023).
Cuturello, F., Celoria, M., Ansuini, A. & Cazzaniga, A. Enhancing predictions of protein stability changes induced by single mutations using MSA-based language models. Bioinformatics 40, btae447 (2024).
Petti, S. et al. End-to-end learning of multiple sequence alignments with differentiable Smith–Waterman. Bioinformatics 39, btac724 (2023).
Lu, W. et al. DynamicBind: predicting ligand-specific protein–ligand complex structure with a deep equivariant generative model. Nat. Commun. 15, 1071 (2024).
Wohlwend, J. et al. Boltz-1 democratizing biomolecular interaction modeling. Preprint at bioRxiv https://doi.org/10.1101/2024.11.19.624167 (2025).
Yu, T. et al. Enzyme function prediction using contrastive learning. Science 379, 1358–1363 (2023).
Luo, F., Wang, M., Liu, Y., Zhao, X.-M. & Li, A. DeepPhos: prediction of protein phosphorylation sites with deep learning. Bioinformatics 35, 2766–2773 (2019).
Nijkamp, E., Ruffolo, J. A., Weinstein, E. N., Naik, N. & Madani, A. ProGen2: exploring the boundaries of protein language models. Cell Syst. 14, 968–978.e3 (2023).
Wang, T. et al. Improved fragment sampling for ab initio protein structure prediction using deep neural networks. Nat. Mach. Intell. 1, 347–355 (2019).
Marchand, A. et al. Targeting protein–ligand neosurfaces with a generalizable deep learning tool. Nature 639, 522–531 (2025).
Ahern, W. et al. Atom level enzyme active site scaffolding using RFdiffusion2. Preprint at bioRxiv https://doi.org/10.1101/2025.04.09.648075 (2025).
Wang, X., Terashi, G., Christoffer, C. W., Zhu, M. & Kihara, D. Protein docking model evaluation by 3D deep convolutional neural networks. Bioinformatics 36, 2113–2118 (2020).
Réau, M., Renaud, N., Xue, L. C. & Bonvin, A. M. J. J. DeepRank-GNN: a graph neural network framework to learn patterns in protein–protein interfaces. Bioinformatics 39, btac759 (2023).
Shuai, R. W., Ruffolo, J. A. & Gray, J. J. IgLM: infilling language modeling for antibody sequence design. Cell Syst. 14, 979–989.e4 (2023).
Montemurro, A. et al. NetTCR-2.0 enables accurate prediction of TCR–peptide binding by using paired TCRα and β sequence data. Commun. Biol. 4, 1–13 (2021).
Lam, J. H. et al. A deep learning framework to predict binding preference of RNA constituents on protein surface. Nat. Commun. 10, 4941 (2019).
Cheng, P. et al. Zero-shot prediction of mutation effects with multimodal deep representation learning guides protein engineering. Cell Res. 34, 630–647 (2024).
Krizhevsky, A., Sutskever, I. & Hinton, G. E. ImageNet classification with deep convolutional neural networks. In Advances in Neural Information Processing Systems (ed Pereira, F. et al.) Vol. 25 (Curran Associates, 2012).
Silver, D. et al. Mastering the game of Go without human knowledge. Nature 550, 354–359 (2017).
Vaswani, A. et al. Advances in Neural Information Processing Systems Vol. 30 (Curran Associates, Inc., 2017).
Radford, A. et al. Learning transferable visual models from natural language supervision. In Proc. 38th International Conference on Machine Learning 8748–8763 (PMLR, 2021).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Vol. 1 (Long and Short Papers) (eds Burstein, J. et al.) 4171–4186 (ACL, 2019).
Zhang, Z. et al. Protein representation learning by geometric structure pretraining. Int. Conf. Learn. Represent. ICLR 2022 (2022).
Wang, Y. et al. Self-play reinforcement learning guides protein engineering. Nat. Mach. Intell. 5, 845–860 (2023).
Lutz, I. D. et al. Top-down design of protein architectures with reinforcement learning. Science 380, 266–273 (2023).
Rumelhart, D. E. & McClelland, J. L. Parallel Distributed Processing: Explorations in the Microstructure of Cognition: Foundations 318–362 (MIT Press, 1987).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
Kipf, T. N. & Welling, M. Semi-supervised classification with graph convolutional networks. Int. Conf. Learn. Represent. ICLR 2017 (2017).
Bronstein, M. M., Bruna, J., LeCun, Y., Szlam, A. & Vandergheynst, P. Geometric deep learning: going beyond Euclidean data. IEEE Signal. Process. Mag. 34, 18–42 (2017).