
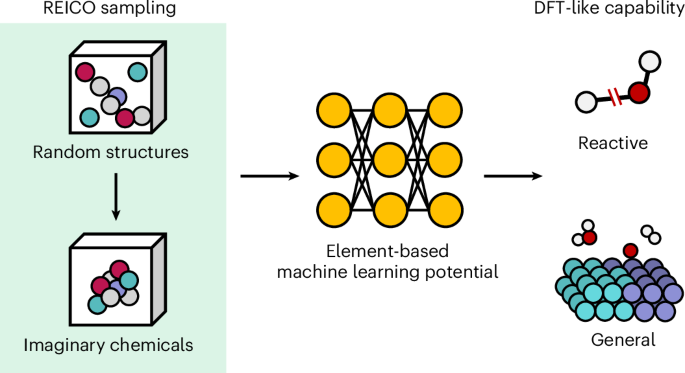
General reactive element-based machine learning potentials for heterogeneous catalysis
作者:Hu, P.
References
Liu, Z.-P. & Hu, P. General rules for predicting where a catalytic reaction should occur on metal surfaces: a density functional theory study of C–H and C–O bond breaking/making on flat, stepped, and kinked metal surfaces. J. Am. Chem. Soc. 125, 1958–1967 (2003).
Alavi, A., Hu, P., Deutsch, T., Silvestrelli, P. L. & Hutter, J. CO oxidation on Pt(111): an ab initio density functional theory study. Phys. Rev. Lett. 80, 3650–3653 (1998).
Oganov, A. R., Pickard, C. J., Zhu, Q. & Needs, R. J. Structure prediction drives materials discovery. Nat. Rev. Mater. 4, 331–348 (2019).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Behler, J. First principles neural network potentials for reactive simulations of large molecular and condensed systems. Angew. Chem. Int. Ed. 56, 12828–12840 (2017).
Zuo, Y. et al. Performance and cost assessment of machine learning interatomic potentials. J. Phys. Chem. A 124, 731–745 (2020).
Huang, S.-D., Shang, C., Zhang, X.-J. & Liu, Z.-P. Material discovery by combining stochastic surface walking global optimization with a neural network. Chem. Sci. 8, 6327–6337 (2017).
Ma, S., Shang, C. & Liu, Z.-P. Heterogeneous catalysis from structure to activity via SSW-NN method. J. Chem. Phys. 151, 050901 (2019).
Wang, H., Zhang, L., Han, J. & E, W. DeePMD-kit: a deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Commun. 228, 178–184 (2018).
Zhang, Y., Xia, J. & Jiang, B. Physically motivated recursively embedded atom neural networks: incorporating local completeness and nonlocality. Phys. Rev. Lett. 127, 156002 (2021).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022).
Batatia, I. et al. The design space of E(3)-equivariant atom-centred interatomic potentials. Nat. Mach. Intell. 7, 56–67 (2025).
Young, T. A., Johnston-Wood, T., Deringer, V. L. & Duarte, F. A transferable active-learning strategy for reactive molecular force fields. Chem. Sci. 12, 10944–10955 (2021).
Smith, J. S. et al. Automated discovery of a robust interatomic potential for aluminum. Nat. Commun. 12, 1257 (2021).
Xu, J., Xie, W., Han, Y. & Hu, P. Atomistic insights into the oxidation of flat and stepped platinum surfaces using large-scale machine learning potential-based grand-canonical Monte Carlo. ACS Catal. 12, 14812–14824 (2022).
Liu, Y. & Guo, H. A Gaussian process based Δ-machine learning approach to reactive potential energy surfaces. J. Phys. Chem. A 127, 8765–8772 (2023).
Nandi, A., Qu, C., Houston, P. L., Conte, R. & Bowman, J. M. Δ -machine learning for potential energy surfaces: a PIP approach to bring a DFT-based PES to CCSD(T) level of theory. J. Chem. Phys. 154, 051102 (2021).
Morrow, J. D., Gardner, J. L. A. & Deringer, V. L. How to validate machine-learned interatomic potentials. J. Chem. Phys. 158, 121501 (2023).
Smith, J. S., Nebgen, B., Lubbers, N., Isayev, O. & Roitberg, A. E. Less is more: sampling chemical space with active learning. J. Chem. Phys. 148, 241733 (2018).
Schran, C. et al. Machine learning potentials for complex aqueous systems made simple. Proc. Natl Acad. Sci. USA 118, e2110077118 (2021).
Shi, Y.-F., Kang, P.-L., Shang, C. & Liu, Z.-P. Methanol synthesis from CO2/CO mixture on Cu–Zn catalysts from microkinetics-guided machine learning pathway search. J. Am. Chem. Soc. 144, 13401–13414 (2022).
Han, Y., Xu, J., Xie, W., Wang, Z. & Hu, P. Comprehensive study of oxygen vacancies on the catalytic performance of ZnO for CO/H2 activation using machine learning-accelerated first-principles simulations. ACS Catal. 13, 5104–5113 (2023).
Han, Y., Xu, J., Xie, W., Wang, Z. & Hu, P. Unravelling the impact of metal dopants and oxygen vacancies on syngas conversion over oxides: a machine learning-accelerated study of CO activation on Cr-doped ZnO surfaces. ACS Catal. 13, 15074–15086 (2023).
Deringer, V. L., Caro, M. A. & Csányi, G. Machine learning interatomic potentials as emerging tools for materials science. Adv. Mater. 31, 1902765 (2019).
Luo, L.-H., Huang, S.-D., Shang, C. & Liu, Z.-P. Resolving activation entropy of CO oxidation under the solid–gas and solid–liquid conditions from machine learning simulation. ACS Catal. 12, 6265–6275 (2022).
Xie, W., Xu, J., Ding, Y. & Hu, P. Quantitative studies of the key aspects in selective acetylene hydrogenation on Pd(111) by microkinetic modeling with coverage effects and molecular dynamics. ACS Catal. 11, 4094–4106 (2021).
Xie, W., Xu, J., Chen, J., Wang, H. & Hu, P. Achieving theory–experiment parity for activity and selectivity in heterogeneous catalysis using microkinetic modeling. Acc. Chem. Res. 55, 1237–1248 (2022).
Deng, B. et al. Systematic softening in universal machine learning interatomic potentials. NPJ Comput. Mater. 11, 9 (2025).
Zeng, J., Cao, L., Xu, M., Zhu, T. & Zhang, J. Z. H. Complex reaction processes in combustion unraveled by neural network-based molecular dynamics simulation. Nat. Commun. 11, 5713 (2020).
Schaaf, L. L., Fako, E., De, S., Schäfer, A. & Csányi, G. Accurate energy barriers for catalytic reaction pathways: an automatic training protocol for machine learning force fields. NPJ Comput. Mater. 9, 180 (2023).
Bernstein, N., Csányi, G. & Deringer, V. L. De novo exploration and self-guided learning of potential-energy surfaces. NPJ Comput. Mater. 5, 99 (2019).
Morrow, J. D. & Deringer, V. L. Indirect learning and physically guided validation of interatomic potential models. J. Chem. Phys. 157, 104105 (2022).
Zhang, S. et al. Exploring the frontiers of condensed-phase chemistry with a general reactive machine learning potential. Nat. Chem. 16, 727–734 (2024).
Chiang, Y. & Benner, P. mace-universal (Revision e5ebd9b). Hugging Face https://doi.org/10.57967/hf/1202 (2023).
Liao, Y.-L., Wood, B. M., Das, A. & Smidt, T. EquiformerV2: improved equivariant transformer for scaling to higher-degree representations. In Twelfth International Conference on Learning Representations (2024).
Chen, C. & Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2, 718–728 (2022).
Hauschild, A. et al. Normal-incidence X-ray standing-wave determination of the adsorption geometry of PTCDA on Ag(111): comparison of the ordered room-temperature and disordered low-temperature phases. Phys. Rev. B 81, 125432 (2010).
Ruiz, V. G., Liu, W., Zojer, E., Scheffler, M. & Tkatchenko, A. Density-functional theory with screened van der Waals Interactions for the modeling of hybrid inorganic–organic systems. Phys. Rev. Lett. 108, 146103 (2012).
Eren, B. et al. Activation of Cu(111) surface by decomposition into nanoclusters driven by CO adsorption. Science 351, 475–478 (2016).
Xu, L. & Mavrikakis, M. Adsorbate-induced adatom formation on lithium, iron, cobalt, ruthenium, and rhenium surfaces. JACS Au 3, 2216–2225 (2023).
Xie, W. & Hu, P. Influence of surface defects on activity and selectivity: a quantitative study of structure sensitivity of Pd catalysts for acetylene hydrogenation. Catal. Sci. Technol. 11, 5212–5222 (2021).
Wu, J., Chen, D., Chen, J. & Wang, H. Structural and composition evolution of palladium catalyst for CO oxidation under steady-state reaction conditions. J. Phys. Chem. C 127, 6262–6270 (2023).
Li, X.-T., Chen, L., Shang, C. & Liu, Z.-P. In situ surface structures of PdAg catalyst and their influence on acetylene semihydrogenation revealed by machine learning and experiment. J. Am. Chem. Soc. 143, 6281–6292 (2021).
Baker, J. & Chan, F. The location of transition states: a comparison of Cartesian, Z-matrix, and natural internal coordinates. J. Comput. Chem. 17, 888–904 (1996).
Cordero, B. et al. Covalent radii revisited. Dalton Trans. https://doi.org/10.1039/B801115J (2008).
Bartók, A. P., Kondor, R. & Csányi, G. On representing chemical environments. Phys. Rev. B 87, 184115 (2013).
Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 134, 074106 (2011).
Thompson, A. P., Swiler, L. P., Trott, C. R., Foiles, S. M. & Tucker, G. J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 285, 316–330 (2015).
Mahoney, M. W. & Drineas, P. CUR matrix decompositions for improved data analysis. Proc. Natl Acad. Sci. USA 106, 697–702 (2009).
Harris, C. R. et al. Array programming with NumPy. Nature 585, 357–362 (2020).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 132, 154104 (2010).
Thompson, A. P. et al. LAMMPS—a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
Steinhardt, P. J., Nelson, D. R. & Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 28, 784–805 (1983).
Menon, S., Leines, G. & Rogal, J. pyscal: a Python module for structural analysis of atomic environments. J. Open Source Softw. 4, 1824 (2019).
Larsen, A. H. et al. The atomic simulation environment—a Python library for working with atoms. J. Phys. Condens. Matter 29, 273002 (2017).
Wang, H.-F. & Liu, Z.-P. Comprehensive mechanism and structure-sensitivity of ethanol oxidation on platinum: new transition-state searching method for resolving the complex reaction network. J. Am. Chem. Soc. 130, 10996–11004 (2008).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Xie, W. Electronic structure calculations. Figshare https://doi.org/10.6084/m9.figshare.29484686.v1 (2025).