
基于序列的基因调控深度学习模型的优化社区努力
作者:de Boer, Carl G.
主界面
在真核生物中,转录因子(TFs)在调控基因表达方面起着关键作用,并且是基因表达组件中的重要组成部分。cis监管机制1,2,3,4,5,6转录因子与核小体和其他转录因子竞争DNA结合,并可以通过生化协同作用相互增强结合,同时它们也会与核小体相互竞争。7,8,9,10尽管该领域在表征调控机制方面取得了重大进展11,12,13,14,15,16,17,18,19,对……的定量理解cis监管仍然是一个主要的挑战。神经网络(NN)在模拟和预测基因调控方面展现了巨大的潜力。尽管不同的网络架构,如卷积神经网络(CNN),11,12,14,19,20循环神经网络(RNNs)21和变压器15,17,18,22,这些模型用于创建基因组模型,但关于神经网络架构和训练策略如何影响其在基因组应用中的性能的研究有限。标准数据集提供了一个共同的基准来评估和比较算法,从而提高了性能并推动了该领域的持续进步。23例如,计算机视觉和自然语言处理(NLP)等领域通过像ImageNet这样的黄金标准数据集的辅助,见证了神经网络的持续改进。23和MS COCO24相比之下,由于基因组学模型通常是为分析特定数据集而临时创建的,因此通常不清楚模型性能的提升是源于更优的模型架构还是更好的训练数据。在许多情况下,所创建的模型与之前的模型无法直接比较,因为用于训练和测试它们的基础数据存在显著差异。
为了应对基因组模型缺乏标准化评估和持续改进的问题,我们组织了随机启动子DREAM挑战赛25在这里,我们要求参与者设计序列到表达模型,并使用具有随机DNA序列的启动子的表达测量数据对其进行训练。这些模型会接收一个调控DNA序列作为输入,并据此预测对应的基因表达值。我们还设计了一组独立的序列来测试模型的极限并提供关于模型性能的见解。在挑战中表现最佳的解决方案超过了之前所有最先进的类似数据模型的表现。我们在各种基准上的评估表明,对于某些类型的序列,模型的性能接近于先前估计的这种类型数据的实验再现性。13而其他模型仍有很大的改进空间。表现最好的模型包括了受实验性质启发的功能以及计算机视觉和自然语言处理领域的最先进模型,并结合了更适合基因组序列数据的训练策略。为了确定个体设计选择如何影响性能,我们创建了一个Prix Fixe框架,该框架能够对单个模型组件进行模块化测试,从而揭示进一步的性能提升。最后,我们在基准测试中使用了表现最佳的DREAM模型果蝇包括从DNA序列预测表达和开放染色质在内的各种数据集,这些模型在所有任务中都超越了现有最先进的模型性能。为了进一步推动该领域的发展,我们将以易于访问的格式提供所有的DREAM挑战赛模型。
结果
随机启动子DREAM挑战和数据集
为了生成竞赛训练数据,我们进行了高通量实验以测量百万随机DNA序列的调控效应(方法先前的研究表明,随机DNA可以表现出与基因组调控DNA相似的活性水平,这是因为偶然包含了多个转录因子结合位点(TFBS)。13,22,26在这里,我们将80 bp的随机DNA序列克隆到启动子样结构中,在黄色荧光蛋白(YFP)上游,将所得文库转入酵母中,在夏多内葡萄汁中培养酵母,并通过流式细胞术(FACS)和测序测量表达。13,27,28 (方法这导致了一个包含6,739,258个随机启动子序列及其相应平均表达值的训练数据集。
我们向参赛者提供了这些数据,他们可以使用这些数据来训练他们的模型,并附有两个关键限制。首先,禁止参赛者以任何形式使用外部数据集,以确保所有模型都基于同一个数据集进行训练。其次,也不允许使用集成预测方法,因为这种方法几乎肯定会提高性能表现,但不会提供关于最佳模型类型和训练策略的任何见解。
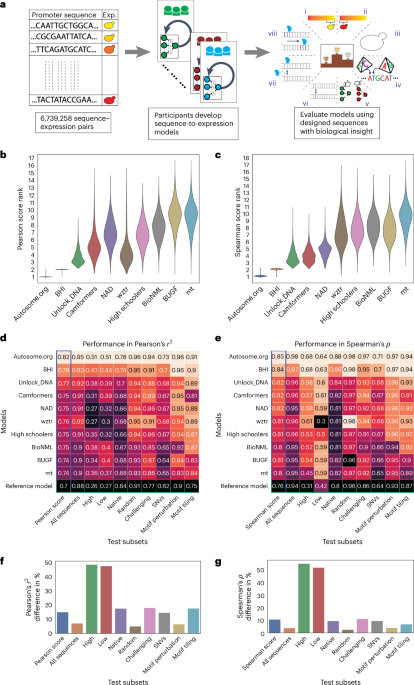
我们在一个旨在从不同角度测试模型预测能力的“测试”序列集合上评估了这些模型。由这些序列驱动的表达水平以与训练数据相同的方式进行量化,但在一个独立实验中对更多的细胞进行了分选(每个序列约100个),这比训练数据测量提供了更准确估计的表达水平,并且在挑战性评估中具有更高的置信度。测试集合包括来自几种启动子序列类型的71,103个序列。我们既包含随机生成的序列也包含了酵母基因组中的序列,以便估算训练领域内的随机序列与自然进化序列之间的性能差异。我们还加入了旨在捕捉先前使用类似数据训练模型已知限制的序列,具体来说是高表达和低表达极端值序列以及设计用于最大化之前开发出的卷积神经网络(CNN)与基于物理学信息的神经网络(“生化模型”)之间预测分歧的序列。13,22我们之前发现,预测紧密相关的序列(即几乎相同的DNA序列)之间表达变化的难度要大得多;因此,我们纳入了模型必须预测由单核苷酸变异(SNVs)、特定转录因子结合位点(TFBSs)的扰动以及在背景序列上TFBSs的排列变化的子集。13,22每个测试子集在评分提交时被赋予了不同的权重,这些权重与集合中的序列数量以及我们认为其重要性如何成比例(表)1例如,预测SNV对基因表达的影响是该领域的一个关键挑战,因为它与复杂性状遗传学密切相关。29因此,在测试集中包含了大量的SNV序列对,并且赋予了SNV最高的权重。在每个序列子集中,我们使用皮尔森相关系数来确定模型性能。r2以及斯皮尔曼的ρ,分别捕获了预测和测量的表达水平(或表达差异)之间的线性相关性和单调关系。在测试子集上每个性能指标的加权总和产生了我们的两个最终性能度量,我们称之为皮尔逊得分和斯皮尔曼得分。
我们的DREAM挑战赛于2022年夏季进行了12周,包括两个评估阶段:公开排行榜阶段和私人评估阶段(图)。1a排行榜在比赛进行到第六周时开放,允许团队每周提交多达20次针对测试数据的预测。在这个阶段,我们使用了13%的测试数据来进行排行榜评估,并仅显示整体Pearson相关系数。r2斯皮尔曼的ρ向参与者提供Pearson得分和Spearman得分,同时保持在促进子集上的表现以及用于评估的特定序列隐藏。参赛团队每周的表现都在提高(扩展数据图)。1),展示了此类挑战在激励更好的机器学习模型开发方面的有效性。来自全球的110多支队伍参加了这一阶段的比赛。在比赛结束时,有28支队伍提交了他们的模型进行最终评估。我们使用剩余的测试数据(约87%)进行了最终评估(图)。1b,c及扩展数据图 fig.2).
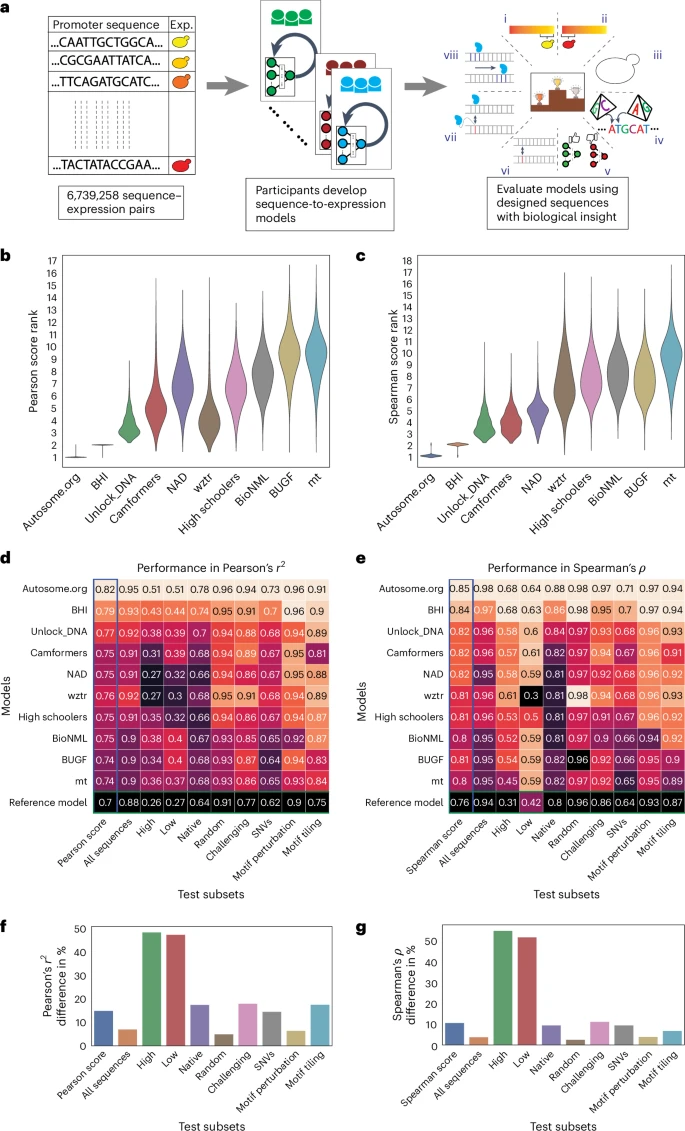
a,左侧,参赛者收到了包含随机启动子及其对应表达值的训练数据集。中间,他们不断优化他们的模型,并在公共排行榜上竞争排名。右侧,在挑战结束时,他们提交了一个最终模型,该模型使用一个测试数据集进行评估,这个测试数据集包含了八种序列类型:(i) 高表达,(ii) 低表达,(iii) 原生,(iv) 随机,(v) 具挑战性,(vi) 单核苷酸变异 (SNVs),(vii) 启动子模体扰动和 (viii) 启动子模体镶嵌。b,c自举法提供了一种对模型预测进行稳健比较的方法。排名分布在n等于10,000个测试数据集中的样本y顶尖团队的轴线) для顶级团队(注意这里的“axes”指的是轴线或坐标轴等概念,在这个上下文中保留了括号内的解释)x皮尔逊得分(轴)b)及斯皮尔曼等级相关系数(c). d,e每个测试数据子集中的最佳团队的表现。每个团队的模型性能(颜色和数值)y轴)在每个测试子集(x皮尔逊相关的轴线)r2 (d)以及斯皮尔曼的ρ (e热图的颜色 palette 按列进行最小值到最大值的归一化。f,g性能差距在最佳模型和最差模型之间被观察到(x轴)在不同的测试子集(y皮尔逊的相关轴)r2 (f)和斯皮尔曼等级相关系数ρ(g百分比差异的计算是相对于每个测试子集的最佳模型性能而言的。
创新的模型设计超越了现有技术水平
我们重新训练了Vaishnav等人提出的变压器模型架构。12之前的最佳性能模型对于此类数据,在挑战数据上进行了测试,并将其作为排行榜上的参考模型(“参考模型”)。顶级提交的整体表现,所有神经网络,都远远优于参考模型。尽管基于注意力的架构近期备受关注22只有挑战前五名提交的作品中有一份使用了Transformer,排名第三。表现最佳的提交作品主要由全卷积神经网络(NN)主导,第一、第四和第五名均由全卷积神经网络获得。表现最好的解决方案基于EfficientNetV2架构。30,31第四种和第五种解决方案基于ResNet架构32而且,所有团队在其模型设计中都以卷积层作为起点。一个具有双向长短期记忆(Bi-LSTM)层的RNN33,34获得第二名。虽然各个团队在许多类似的训练策略上大致趋于一致(例如使用Adam优化器35或AdamW36(优化器而言),它们也有很大的差异(表)2).
参赛队伍提出了几种创新的方法来解决表达预测问题。表现最好的队伍Autosome.org通过训练网络预测表达区间概率向量,将其平均以估算表达水平,从而将任务转化为软分类问题,这种方法有效地重现了实验中数据的生成方式。他们还使用了一种不同的数据编码方法,在传统的四通道DNA序列的一热编码(OHE)基础上增加了两个额外的通道:一个通道表示输入序列是否可能仅在一个细胞中测量(这会导致整数表达值),另一个通道表示输入序列是否以反向互补方向提供。此外,Autosome.org模型仅有200万个参数,在前十名提交的作品中参数最少,证明了有效设计可以大大减少所需的参数数量。Autosome.org和BHI在训练最终模型时使用整个提供的训练数据(即没有保留用于验证的序列),并在预先确定的周期数内进行训练(通过交叉验证使用验证子集来决定)。排名第三的队伍Unlock_DNA采用了一种新颖的方法,随机屏蔽输入DNA序列中的5%,然后让模型预测被屏蔽的核苷酸和基因表达。这种方法将被屏蔽核苷酸的预测用作正则化器,并向模型损失函数添加了重建损失,从而稳定了其大型神经网络(NN)的训练过程。排名第9的队伍BUGF采用了一种类似但略有不同的策略:他们随机突变了序列中的15%,并计算了一个额外的二元交叉熵损失来预测序列中是否有任何碱基对被突变。排名第五的团队NAD使用了GloVe37为了每个基础位置生成嵌入向量,并将这些向量用作其神经网络的输入,而其他团队则使用传统的OHE DNA序列。两个团队,SYSU-SAIL-2022(第11名)和Davuluri实验室(第16名),尝试训练DNA语言模型38通过预训练一个BERT(变压器中的双向编码器表示)语言模型来对挑战数据进行处理。39在挑战数据上进行了测试,并使用BERT嵌入训练了一个表情预测器。
测试序列子集揭示模型差异
对不同测试子集上模型性能的分析揭示了不同模型面临的独特和共同挑战。前两名模型在每个测试子集中无论评分指标如何都排名第一或第二(有时并列),表明它们的优越表现不能归因于任何单一的测试子集(图.)1天,e此外,每个测试子集内的排名有时会在皮尔森得分和斯皮尔曼得分之间有所不同,这进一步证明了这两种衡量标准以不同的方式捕捉性能(图。)1天,e).
尽管对于随机序列和本地序列模型的排名相似,但与随机序列相比,本地酵母序列中模型性能的差异更大。具体来说,模型之间的表现差距在本地序列为高达17.6%,而在随机序列为仅5%(皮尔逊相关系数的)r2图。1f类似地,对于斯皮尔曼等级相关系数,这一差异为9.6%(本地)对2.7%(随机)ρ(图。)1克这表明顶级模型学习了更多进化产生的调控语法。此外,原生序列和随机序列在性能上的显著差异表明还有更多的调控逻辑有待学习(尽管原生DNA的序列覆盖率较低,可能是因为其重复内容较高,从而降低了数据质量和该组的预测能力;扩展数据图)。3).
模型在准确预测基因表达极端范围内的变异方面也表现出高度的差异。细胞分选器在最低表达水平下的信号与噪声比降低,并且分选箱的位置可能会截断表达分布的尾部。6,12总体而言,在这些子集中各团队的模型表现差异最大,表明挑战模型在不同程度上能够克服这些问题。例如,皮尔逊相关的中位数差异为r2高性能组和低性能组之间的差异约为48%,而对于其他组则是16%(图。)1f,g).
模型在预测紧密相关的序列之间表达差异的能力也有所不同(图。)1de,‘SNV’,和扩展数据图 figs.4以及5),模型性能在更细微的变化中差异更为显著。具体来说,在皮尔森相关系数中的最佳和最差百分比差异为r2以及斯皮尔曼的ρ对于motif扰动分别为6.5%和4%,对于motif平铺分别为17.7%和7%,对于SNV分别为14.6%和9.6%,这表明表现最佳的模型更好地捕捉到了这些细微差别CIS-法规。这与我们对影响微妙性的理解一致;扰乱TFBS(基序扰动,即我们强烈匹配重要转录因子的同源基序的位置进行序列突变或改变结合位点的数量)代表了一种相对较大的干扰,并且可以用简单的模型来预测这些模型可以捕捉到这些转录因子的结合并计算TFBS实例。然而,当TFBS在背景序列上排列时,每个序列中都存在相同的TFBS,模型必须学会其位置如何影响其活性,除了捕获所有由于基序排列而创建或破坏的次级TFBS之外。13最后,SNV(单核苷酸变异)甚至更难预测,因为序列几乎完全相同,只是在一个核苷酸上有所不同,而这可能以微妙的方式影响多个转录因子的结合。
固定价格框架揭示了最优模型配置
DREAM挑战赛中排名前三的解决方案不仅因为其性能相比其他模型有显著提升而脱颖而出,还因其在数据处理、预处理、损失计算以及多样化的神经网络层(包括卷积、递归和自注意力机制)方面的独特方法而著称。为了识别影响它们性能的因素,我们开发了一个Prix Fixe框架,该框架将每个解决方案分解为不同的模块,并通过选择每种类型的单个模块来测试来自每个解决方案的任意组合的模块(图。2a我们在该框架内重新实现了前三种解决方案,并发现81种可能的组合中有45种是兼容的。我们移除了每个解决方案独有的、无法跨方案比较的特定测试时间处理步骤。最后,我们将所有兼容的组合使用相同的训练和验证数据进行了再训练,解决了某些原始解决方案曾使用整个数据集进行训练的问题。我们的方法促进了对不同组件对整体性能的个体贡献进行系统且公平的比较。

a该框架将每个团队的解决方案分解为模块,使得不同解决方案的模块可以被组合在一起。bPearson得分的性能是从 Prix Fixe 对来自DREAM挑战赛前三名解决方案的所有模块组合进行评估的结果。每个单元格代表由核心层块(主要行,左侧)、数据处理器和训练器(主要列,顶部)、第一层块(次要行,右侧)以及最终层块(次要列,底部)模块的独特组合所获得的性能。灰色单元格表示在训练过程中不兼容或未收敛的组合。c性能(皮尔逊得分,)y三个数据处理器和训练模块的轴)x包括各自的模块(个别点)在内的每个固定价格模型的轴和颜色。原始模型组合用白色点表示,而所有其他组合用黑色点表示。d参数数量(x对于DREAM挑战赛的前三种模型(Autosome.org、BHI和UnlockDNA),以及它们基于核心层块的最佳表现对应模型(DREAM-CNN、DREAM-RNN和DREAM-Attn)在Prix Fixe运行中的结果轴(result axis)。y轴).e, 如同d但显示每个模型的皮尔森得分(x轴).
我们的分析揭示了Autosome.org卓越表现的来源以及不同模型组件之间的相互作用,以及它们进一步优化的潜力。当使用Autosome.org的数据处理器和训练器重新训练BHI和UnlockDNA NN时,它们的表现有了显著提升(图。)2b,c及扩展数据图(figs.)6以及7此外,每个团队的模型架构都可以进一步优化,从而实现性能更好的模型(图)。2c使用相同的骨干模块但参数类似或更少(图。)2D然而,除了Autosome.org的数据处理器和训练器模块之外,没有其他模块组件占据主导地位,它们的性能似乎取决于与其他哪些模块组合使用(补充图)。1对于Autosome.org的每个核心模块BHI和UnlockDNA,我们将最优的Prix Fixe模型分别命名为DREAM-CNN、DREAM-RNN和DREAM-Attn。DREAM模型学习到了非常相似的观点的表示。CIS监管逻辑如相似的归因分数所示(扩展数据图)8使用计算模拟诱变(ISM)。有趣的是,除了在可识别的共识转录因子结合位点(TFBS)被改变时同意其显著影响外,模型还一致地预测了较小的影响,这些影响在1-3 bp范围内符号变化,而这个长度太短无法对应于共识TFBS。40,支持这样的观点,即低亲和力结合位点的丰富性在许多方面扮演着重要角色cis调控元件(CREs)7,13,41.
优化后的模型在其他物种和数据类型上超过了当前最佳性能
为了确定我们在酵母数据上优化的模型架构和训练策略是否可以应用于其他物种,我们接下来将它们应用到了果蝇(黑腹果蝇)并在一系列多样化的任务上进行了测试。首先,我们测试了它们预测基因调控活性的能力,这些活性是在D. 黑腹果蝇在自我转录活性调节区域测序(STARR-seq)大规模并行报告基因检测(MPRA)的背景下(涉及发育和管家型启动子)。这从根本上代表了模型旨在解决的序列到表达的问题,尽管是在不同的生物体中。果蝇与酵母相比),实验测量方法(RNA测序与细胞分选),更长的序列(249 bp 与 150 bp),较小的数据集(约50万 对比 670万)以及从单一任务到多任务框架的转变(两种启动子类型)。我们比较了DREAM优化模型和DeepSTARR42基于Basset的最先进的CNN模型20架构特别开发用于预测我们在此基准测试中使用的数据(带有唯一分子标识符集成的STARR-seq(UMI-STARR-seq))43在D. 黑腹果蝇S2细胞42,44为了进行稳健的比较,我们使用交叉验证训练模型,并始终在相同的保留测试数据上进行评估。方法我们的模型在发育和管家转录程序上都持续优于DeepSTARR(图示)。3a),DREAM-RNN的模型性能超过了DREAM-CNN和DREAM-Attn。

a, D. 黑腹果蝇STARR-seq42预测。对于预留数据,预测的增强子活性与实际增强子活性之间的皮尔逊相关系数(Pearson's correlation)y(轴)用于两个不同的转录程序(x每个模型(颜色)的轴。b人类MPRA45预测。针对保留数据集,预测表达与实际表达之间的皮尔逊相关系数(Pearson correlation for predicted versus actual expression for held-out data)y(三个不同的哺乳动物细胞类型)的MPRA数据集的轴线)x每个模型(颜色)的轴。c,d人类可访问性(批量K562 ATAC-seq)46,49预测。对于每个模型(x轴和颜色),模型性能(y轴以皮尔逊相关系数的形式表示,该系数用于预测与实际读取计数之间的关系(每元素)c)以及预测与实际染色质可及性谱型在每个元素上的1-中位数Jensen-Shannon距离d). 在a–d,点代表交叉验证的折数,性能是在保留的测试数据上进行评估的P由……决定的值t-对照最先进的模型与优化后的模型进行的配对双侧检验结果如模型性能分布图上方所示。e参数数量的比较x用于染色质可及性预测任务的不同模型的轴。
为了进一步验证我们的模型的普适性,我们接下来使用基于 lentivirus 的 MPRAs(lentiMPRAs)训练了 DREAM 优化后的模型,这些 MPRA 在三种人类细胞类型中测试 CRE:肝细胞(HepG2)、淋巴母细胞(K562)和诱导多能干细胞(WTC11)。45在这里,我们的模型需要从更小的数据集(约56,000至226,000 vs 6.7百万)中捕捉更为复杂的调控活动。我们将这些模型与MPRAnn进行了比较。45,一个针对这些特定数据集优化的CNN模型(方法所有模型均采用交叉验证进行训练,并像最初训练MPRAnn时一样在独立的测试数据上进行评估。45经过DREAM优化的模型在性能上远远超过了MPRAnn,而且随着训练数据的增多,两者之间的性能差距进一步扩大(图)。3b唯一的例外是DREAM-Attn,在最小的数据集(WTC11;56,000个序列)上没有超过MPRAnn。同样,DREAM-RNN在我们的模型中表现出最佳性能,特别是在较大的数据集上。
为了在一个与CRE功能相关的独立预测任务上评估模型,我们在预测开放染色质的任务上评估了我们的优化模型。具体来说,我们将我们的优化模型与ChromBPNet进行了比较。46,47,48基于BPNet的16一种用于预测转座酶可访问染色质测序(ATAC-seq)信号的模型,该模型针对开放染色质区域进行。在这里,输入的DNA序列长度约为酿酒酵母启动子上优化DREAM模型长度的14倍(2,114 bp 对比 150 bp),并且这些模型现在需要同时预测一个中心1,000-bp片段的整体可访问性(读取计数)和可访问性分布情况,而不是预测单一表达值。由于如此长的输入序列导致注意力模块所需的内存过大,无法训练DREAM-Attn模型,但我们对其他优化后的DREAM模型以及ChromBPNet在K562批量ATAC-seq数据上进行了训练和评估。49 (方法DREAM-RNN 在读数计数和染色质可及性预测上都显著优于ChromBPNet(图)。3c,d),强调了我们的模型甚至在差异很大的场景下也具有适应性。cis监管数据类型。相比之下,DREAM-CNN的表现与ChromBPNet相当46读计数预测(图。)3C)但在预测染色质可及性谱型方面效果较差(图。3D).
值得注意的是,这些评估中DREAM优化模型的架构和训练范式几乎没有改变(扩展数据图。9无法容纳数据的组件被丢弃(例如,表示单例观测值的输入编码通道与STARR-seq、MPRA和ATAC-seq数据不兼容;方法唯一的其他修改是为了让预测头能够预测新的任务(例如,最终层块的架构和使用特定于任务的损失函数;方法)或适应于相对于DREAM数据集较少的训练序列数量(减少批量大小和/或最大学习率(LR);方法值得注意的是,在所有这些次要基准测试中,DREAM-RNN 的表现优于其他 Prix Fixe 优化的模型(图 Fig.)3a–d),突出其优秀的泛化能力。
讨论
2022年随机启动子DREAM挑战赛为参与者提供了一个独特的机会,提出用于建模调控序列的新颖模型架构和训练策略。参赛者在数百万个随机调控DNA序列及其相应的表达测量数据上训练了序列到表达的模型。一组设计好的序列被用来评估这些模型并测试它们的极限。值得注意的是,DREAM挑战赛中的19个模型超越了之前的最佳状态。22(扩展数据图 fig.)4),其中大多数使用了独特的架构和训练策略。为了系统地分析模型设计选择对其性能的影响,我们开发了Prix Fixe框架,在该框架中将模型抽象为模块化部分,使我们能够结合来自不同提交的模块以识别对模型性能的关键贡献者。我们将Prix Fixe框架应用于挑战赛中排名前三且其神经网络架构(CNN、RNN 和自注意力)和训练策略差异显著的模型,并在每种情况下都能构建出改进后的模型。
神经网络的训练策略对其模型性能的影响与网络架构本身一样显著(图。)2c及扩展数据图(fig.)7在固定价格赛跑中,通过使用软分类来训练网络预测表达的分布而不是精确值,有助于模型捕捉更多的信息。cis这些发现表明,不仅应该关注网络架构,还应注重训练过程的优化和预测任务的重新定义,以保持平衡的重点。
值得注意的是,DREAM挑战赛中表现最佳的模型表明,如果优化得当,参数较少的简单神经网络架构可以有效捕捉单个CREs的大部分活动。在前五名提交的作品中,有三个没有使用变压器,包括表现最好的团队(其使用的参数也是前十中最少的)。利用我们的Prix Fixe框架,我们成功设计了与对照模型相比不仅包含相似或更少的参数,而且性能更优的模型(图)。2d,e此外,这些经过DREAM优化的模型在其他任务上始终优于之前的最先进模型。cis监管任务,尽管其参数与之前的模型相比相当(甚至更少)(图示)。1天以及3e及扩展数据图(fig.)10). 虽然基因组模型设计需要考虑任务的性质(例如,增强子-基因调控必然要求能够捕获长程互作的能力),我们的研究结果表明,构建更好的模型主要依赖于有效的优化而不是简单地增加模型容量。然而,随着生物化学以更高分辨率进行近似,构建更好的模型可能会带来计算负担的增加(扩展数据图)。10).
在DREAM挑战中,我们观察到测试子集间的结果各异,这说明了评估的复杂性。cis有效地实施监管模型。例如,对于与训练数据(也是随机序列)在同一领域的随机序列的性能表现相对一致(图。)1天,e相反,将领域转移到原生序列上则凸显了模型之间的差异,因为各种调控机制的相对频率可能不同,这是由于它们的进化起源所致(图)。1天,e这表明一个在整体建模方面表现出色的模型CIS监管可能在涉及某些调控机制(例如,进化序列中的协同作用)的序列上表现不佳,这些机制难以从训练数据中学习,导致对使用这些机制的序列的生化机制和变异效应预测不准确。这强调了对基因组模型进行全面评估的重要性。50并设计特定的数据集来测试这些模型的极限。
为了持续改进基因组学模型,需要标准化、稳健的基准数据集。DREAM挑战数据集解决了这一需求,并通过展示DREAM优化模型在不同情境下的通用性,证明了此类标准化数据集所能产生的影响。果蝇并且在无需额外模型调优的情况下,这些模型和任务可以应用于人类数据集。然而,值得注意的是,来自这一挑战的模型仅探索了可能的设计空间的一小部分,并且很可能会得到改进。此外,通过调整这些模型的超参数以适应特定的数据集或使用这些模型的集成,可以针对不同的数据集优化DREAM优化模型的性能。我们提供的数据集以及Prix Fixe框架作为一个宝贵的资源,用于继续探索和开发专门针对DNA序列的创新神经网络架构和训练方法。此外,由于DREAM优化模型的模块化特性和已证明的泛化能力,其他研究人员可以轻松地将这些模型应用于其他基因组学问题。
方法
设计测试序列
高表达和低表达序列是使用DEAP设计的58将变异概率和两点交叉概率设置为0.1,选择锦标赛规模为3,初始种群大小为100,000,并运行十代遗传算法,使用在随机酵母启动子序列上训练的CNN的预测作为适应度函数22原生测试子集序列是通过将原生酵母启动子分割成80 bp的片段来设计的。13从之前的实验中采样了随机序列,在该实验中测试的DNA是随机合成的(就像训练数据一样)并进行了定量分析。13挑战序列是通过最大化由CNN模型预测的表情之间的差异来设计的。22以及一个生化模型(一种物理信息神经网络)13这些序列代表了在使用DEAP库中的遗传算法优化每一代100个序列的种群时,模型之间表达差异的帕累托前沿。该遗传算法具有0.02的每碱基突变率和0.5的重组率,并且进行了100代的进化。58以及一个自定义脚本(GASeqDesign.py)59). Vaishnav等人表示的大多数SNV代表了序列轨迹。22但还包括添加到随机、设计和天然启动子序列中的随机突变。基序扰动包括Reb1和Hsf1的扰动。具有扰动的Reb1结合位点的序列通过插入Reb1共识结合位点(强或中等亲和力;正向和反向互补方向)创建,然后在每个基序出现位置添加1-3个SNV,并将规范和突变的基序出现在位置20或80处嵌入到十个随机生成的序列中。具有Hsf1基序出现的序列通过使用包含1-10个单体共识位点(ATGGAACA)的随机背景序列进行平铺设计,这些位点从随机起始序列的左右两侧依次添加,并分别在可能的八个位置内单独插入或类似地以平铺方式插入1-5个三聚体Hsf1共识位点(TTCTAGAANNTTCT)。基序平铺测试子集序列通过将每个基序的单个规范(poly(A),AAAAA;Skn7,GTCTGGCCC;Mga1,TTCT;Ume6,AGCCGCC;Mot3,GCAGGCACG;A zf1,TAAAAGAAA)嵌入到三个随机生成的背景序列中的每个可能位置和方向中来设计(基序完全包含在80 bp可变区域中)。13.
定量启动子表达
高复杂度的随机DNA文库作为训练数据创建,使用吉布森组装将双链随机DNA片段嵌入到双报告载体酵母_DualReporter(AddGene, 127546)中。该随机DNA片段通过退火互补引物序列并利用Phusion聚合酶混合试剂盒(New England Biolabs)延伸成双链,然后进行凝胶纯化后再克隆。随机DNA被插入到远端(GCTAGCAGGAATGATGCAAAAGGTTCCCGATTCGAACTGCATTTTTTTCACATC)和近端(GGTTACGGCTGTTTCTTAATTAAAAAAAGATAGAAAACATTAGGAGTGTAACACAAGACTTTCGGATCCTGAGCAGGCAAGATAAACGA)启动子区域之间。随机启动子库在大肠埃希菌理论上包含约7400万个随机启动子(通过稀释和平板培养估计),并转入S288c中Δura3产生2亿个转化子的酵母,在SD-Ura培养基中进行了筛选。然后,将该池接种到1升过滤后的夏多内葡萄汁中,以在600 nm处开始产生光密度(OD)600初始OD值为0.05,在室温下培养,无需持续振荡。根据需要使用新鲜霞多丽葡萄汁进行稀释以保持OD值低于0.4,总培养时间为48小时,并经历了超过5代的生长。在每次测量OD值之前,轻轻摇晃培养物以去除碳酸化,等待泡沫消散后再摇晃直至不再有气泡释放。分选前,酵母被离心沉降,用冰冷的PBS洗一次,再悬浮于冰冷的PBS中,在冰上保存,然后通过log进行分选。2在贝克曼库尔特MoFlo Astrios上使用mCherry和绿色荧光蛋白的吸收和发射检测红色荧光蛋白(RFP)/黄色荧光蛋白(YFP)信号(使用受pTEF2调控的组成型RFP来控制外源噪声)13细胞被分成了18个均匀的组,每三批六个组。排序后,每个组中的细胞被离心并重新悬浮在SC-Ura中,然后在30°C下振荡培养2-3天。提取质粒,扩增启动子区域,并通过PCR添加Nextera接头和多重索引序列,生成的文库随后进行测序,在Illumina NextSeq上使用150个循环套件的2×76 bp成对末端读数合并测序文库并测序。设计(测试)实验类似地执行,但文库是从Twist寡核苷酸池通过PCR扩增获得的。E. coli转换复杂度仅为105,覆盖库的范围超过10倍。
为了获得随机启动子序列的序列-表达配对,使用中间重叠的序列将代表启动子序列两侧的双端读段进行比对,要求重叠部分为40±15 bp,并丢弃未能在这些限制内良好比对的任何读段。13为了将相关的启动子序列合并为一个代表序列,我们使用Bowtie2(参考文献)将其在每个库中观察到的序列相互对齐。60),创建一个包含实验中观察到的所有唯一序列的Bowtie数据库(默认参数),并对其进行比对,这些相同的序列允许多映射读段(参数包括‘-N 1 -L 18 -a -f -no-sq -no-head -5 17 -3 13’)。任何相互比对上的序列都被分配到同一个簇中,并使用每个簇中最具有读数的序列为该簇的真实启动子序列。对于每个启动子序列,其表达水平被估计为观察到该启动子的区间加权平均值。13对于设计的(测试)库,我们直接将读段与我们订购的序列的Bowtie数据库进行比对以量化,并使用MAUDE估计它们的表达水平。61以每个分类bins中的读取丰度作为输入,并估计每个序列的初始丰度为其在所有bins中的平均相对丰度。
竞赛规则
-
1.
只有提供的训练数据可以用于训练模型。模型必须从零开始训练,不得在外部数据集上进行预训练,以避免对测试数据中存在的序列过度拟合(例如,测试数据中的一些序列源自现有的酵母启动子)。
-
2.
可重复性是所有提交的先决条件。参赛者必须提供代码和重现其模型的说明。我们重新训练了最佳解决方案以验证它们的表现。
-
3.
增加提供的训练数据是允许的。对提供的测试数据进行伪标签是不允许的。在训练过程中以任何目的使用测试数据都是不允许的。
-
4.
不允許集會。
有关竞赛和其指南的详细信息可以在DREAM挑战网页上找到。https://www.synapse.org/#!Synapse:syn28469146/wiki/617075).
性能评估指标
我们计算了皮尔逊的r2以及斯皮尔曼的ρ在每个序列子集的预测值和测量值之间。各个性能度量指标在启动子类型上的加权总和产生了我们最终的两个性能度量标准,我们称之为Pearson得分和Spearman得分。
$${\rm{皮尔逊}}\;{\rm{得分}}=\frac{\mathop{\sum }\limits_{i=0}^{{子集数}}{{w}_{i}}\times {\rm{皮尔逊}}\;{{r}^{2}}_{i}}{\mathop{\sum }\limits_{i=0}^{{子集数}}{{w}_{i}}}$$
斯皮尔曼得分=\(\frac{\sum_{i=0}^{\text{subsets}} w_i \times \text{Spearman}_i}{\sum_{i=0}^{\text{subsets}} w_i}\)
这里,wi是用于的权重i测试子集(表1). 皮尔逊\(r^2_i\)以及斯皮尔曼系数_i_分别是皮尔逊系数和斯皮尔曼系数的平方,用于序列间的i该子集。
自举分析模型性能
为了确定模型的相对性能,我们进行了自助法分析。在这里,我们10次抽取了测试数据的10%,并为每个样本计算了每个模型以及基于皮尔逊和斯皮尔曼评分的模型排名。我们将两个指标的排名平均值来决定它们的最终排名。
参赛者使用的方法描述
挑战参与者所采用的方法概述如下:补充信息.
固定价格框架
Prix Fixe框架,用Python和Pytorch实现,通过将整个过程模块化(从数据预处理到预测),并强制执行特定的输入和输出格式以允许来自不同方法的组件集成,从而简化了神经网络的设计和训练。以下描述了Prix Fixe框架中的各个模块。
数据处理和训练人员
数据处理器类专用于将原始DNA序列数据转换为可用于后续神经网络训练的可用格式。数据处理器可以生成一个可迭代对象,提供包含特征矩阵的字典x“(输入到神经网络)和目标向量‘y‘’(预期输出)。可以包含额外的键以支持扩展功能。此外,数据处理器可以提供启动NN块所需的基本参数,例如确定第一层中的通道数。
训练器类管理神经网络的训练。它处理来自数据处理器的数据批次,并将它们输入到神经网络中。如果需要,它会计算辅助损失,与主损失一起从神经网络中获取,从而在训练过程中简化复杂的损失计算。
固定价格净价菜单
此模块包含了整个神经网络架构:
-
(i)
第一层模块:这构成了网络的原始层。它们可能包括初始卷积层或实现特定编码机制等功能,如k输入的编码方式为UTF-8。注意,由于给定的信息较少,“-mer”部分无法准确翻译或理解其具体含义,在没有更多信息的情况下,保持原样。如果“-mer”是特定上下文中的缩写或者有特殊含义,请提供更多的信息以便更精确地翻译。原文:-mer encoding for the input.
-
(ii)
核心层模块:这代表中央架构组件,包含残差连接、LSTM机制和自注意力等元素。该模块的模块化构造也允许灵活组合,例如堆叠一个残差CNN块与一个自注意力块。
-
(iii)
最终层块:此阶段通过使用如池化、展平和全连接层等层来缩小潜在空间,以产生最终预测。它计算预测结果,并与损失一同输出。
对于这三个模块,标准的输入格式是(批量大小,通道数,序列长度)。前两个模块以一致的格式输出(批量大小,通道数,序列长度),而最后一个模块提供预测的表达值。每个模块都可以传播自身的损失。整个框架是在PyTorch中实现的。
为了确保在Prix Fixe框架中各解决方案之间的公平比较,我们去除了每个解决方案特有的测试时间处理步骤。我们将DREAM Challenge数据集划分为两个部分,分配90%的序列用于训练,10%用于验证。使用这些数据,我们在框架内重新训练了所有兼容的组合。在81种潜在组合中,我们确定有45种是兼容的,并且其中41种成功完成了训练过程。由于图形处理单元(GPU)内存限制,我们调整了一些组合的批处理大小。
Prix Fixe平台上的DREAM优化模型运行结果
数据处理和训练人员
启动子序列在5′端通过质粒中的恒定片段扩展,标准化为150 bp的长度。这些序列经过OHE转换为四维向量。“单例”启动子,在所有bin中仅出现一次,被归类为整数表达估计值。考虑到这些单例表达估计值可能存在变异性,引入了一个二进制“is_singleton”通道,对于单例标记为1,否则标记为0。为了考虑基于相对于转录起始位点的链取向的不同调控元件的行为差异,在训练集中每个序列都以原始形式和反互补形式提供,并通过“is_reverse”通道(原序为0,反序为1)进行标识。因此,输入维度设置为(batch, 6, 150)。
该模型的训练使用了AdamW优化器,并设置了weight_decay为0.01。对于大多数模块,最大学习率(LR)被设置为0.005,而对于注意力块,则采用了更为保守的学习率为0.001,这是因为自注意力机制对较高学习率较为敏感。该学习率按照一个周期性学习率策略进行调度。62,该训练过程分为两个阶段,并采用了余弦退火策略。训练数据被分割成每批大小为1,024的批次,整个训练过程历时80个纪元。模型的性能和选择依据是Pearson相关系数最高的标准。r验证数据集中观察到的值。
在预测过程中,数据处理与数据处理器相同,只是将‘is_singleton’设置为0。然后对原始序列和反向互补序列的预测结果进行平均。
固定价格净价菜单
DREAM-CNN
第一层块:OHE输入通过一维(1D)CNN进行了处理。借鉴了DeepFam的思想63使用了卷积层,其核大小分别为9和15,这与ProSampler识别的常见基序长度相呼应。64每一层的通道大小为256,使用修正线性单元激活函数,并包含0.2的dropout率。这两层的输出沿通道维度进行了拼接。
核心层模块:该段包含六个卷积块,其结构受EfficientNet架构的影响。该段进行了修改,例如将深度可分离卷积替换为分组卷积,并使用了挤压和激励(SE)块。65并采用通道级拼接方式进行残差连接。通道配置从初始块的256个通道开始,随后依次为128、128、64、64、64和64个通道。66.
最终层块:最后一个块由一个逐点卷积层组成,后面跟着通道方向上的全局平均池化和SoftMax激活函数。
DREAM-RNN
第一层块:与DREAM-CNN相同。
核心层块:使用了双向LSTM(Bi-LSTM),设计用于捕捉基序依赖性。LSTM的隐藏状态每个维度为320,拼接后结果为640维。随后加入了一个类似第一层块的CNN块。
最终层块:与DREAM-CNN相同。
DREAM-Attn
第一层块:这一段是一个标准的卷积层,卷积核大小为7,随后是BatchNorm操作67并使用sigmoid线性单元激活函数68.
核心层模块:此模块使用了Proformer69,一种类似于马卡龙的变压器编码器架构,在编码器块的开始和结束处使用两个半步前向网络(FFN)层。此外,在初始的FFN层之后和多头注意力层之前集成了一个可分离的一维卷积层。
最终层块:与DREAM-CNN和DREAM-RNN相同。
人类MPRA模型
在每个大规模的MPRA库中,每一个序列及其对应的反向互补序列都被分组在一起,并且这些成对的序列被分配到十个不同的交叉验证折集中,以确保正向和反向序列都位于同一折集中。DREAM-CNN、DREAM-RNN、DREAM-Attn 和 MPRAnn 使用这十个折中的九个进行训练,保留一个折用于评估模型性能。对于每个独立的测试折集,使用九个模型进行训练,其中一个折专门用于验证目的,其余八个作为训练折集。来自这九个模型的后续预测被汇总起来,平均值用作独立测试数据的最终预测。
MPRAnn架构45使用学习率为0.001,带有耐心为10的早期停止标准,在100个周期内,批量大小为32以及均方误差损失函数的Adam优化器进行训练。对于DREAM-CNN、DREAM-RNN和DREAM-Attn,那些无法容纳Agarwal等人数据的部分被舍弃。例如,“is_singleton”通道对于MPRA数据无关紧要,并且由于将问题转换为软分类不可行,因此使用均方误差(而非Kullback–Leibler散度)来进行损失计算。MPRAnn使用的批量大小比我们的DREAM优化训练器小得多(32与1,024相比),所以我们将其调整为与MPRAnn相同。没有对模型结构或训练范式进行其他更改。
果蝇UMI-STARR-seq模型
DeepSTARR架构42使用学习率为0.002,具有10个周期的早期停止标准,在100个周期内,批量大小为128以及均方误差损失函数的Adam优化器进行了训练。对于DREAM-CNN、DREAM-RNN和DREAM-Attn,我们使用了与人类MPRA数据集相同的设置,但输出层被修改为预测与管家基因和发育启动子相关的两个值,并且损失计算为每个输出的均方误差之和(如同DeepSTARR)。
仅五大最大的果蝇染色体(Chr2L、Chr2R、Chr3L、Chr3R和ChrX)用作测试数据。对于每个独立的测试染色体,其余序列被分配到十个不同的训练-验证折叠中,并分别训练了DREAM-CNN、DREAM-RNN、DREAM-Attn和DeepSTARR模型(每种十个)。随后,从这十个模型中聚合预测结果,平均值作为独立测试染色体的最终预测。
人类染色质可及性模型
我们使用了之前研究中提出的五个独立的训练集-验证集-测试集划分46针对人类K562细胞系的ATAC-seq实验。对于这些分区中的每一个,我们首先训练了五个不同的偏差模型,每个折叠一个,旨在捕捉存在于ATAC-seq谱型中的由酶驱动的偏差。随后,使用相同的偏差模型对ChromBPNet、DREAM-CNN和DREAM-RNN在每个折叠中进行了训练。对于DREAM-CNN和DREAM-RNN,采用从ChromBPNet预测头(1D卷积层、裁剪层、平均池化层和密集层)用于最终层块以预测可访问性谱型和读取计数。输入编码通道is_singleton和is_rev被省略。在这种情况下,我们修改了DREAM优化的训练器,使其使用与ChromBPNet相同的批次大小(从1,024减少到64),并减少了最大学习率(从5 × 10−3到5×10−4没有对模型的结构或训练范式进行其他任何修改。
对于此任务,我们在TensorFlow中重新实现了DREAM-CNN和DREAM-RNN架构,以确保所有模型具有相同的偏差模型。这种方法选择的代价是不得不将某些组件(输入编码、AdamW优化器等)排除在经过DREAM优化的数据处理器和训练器之外。然而,这保证了模型之间的一致性,从而实现了不同架构之间的无偏比较。
ISM
ISM得分是通过为每个DNA序列创建所有可能的单核苷酸突变并计算相对于原始序列预测表达的变化来获得的。然后,通过在该位置上平均各个核苷酸的突变分数,确定了每个位置上的单一ISM得分。
每批次平均训练时间和吞吐量
我们测量了使用批大小为64的人类ATAC-seq数据集以及另外两个批大小为32的数据集的训练时间。通过测量每个模型每秒可以预测多少数据点(不包括反向传播)来确定吞吐量。从批大小32开始,我们将批大小逐步加倍(64、128、256等),并在每个阶段记录吞吐量,直到达到GPU支持的最大批大小为止。我们为每个模型处理了5000个批次以计算平均每个批次的训练时间,并且为了测量吞吐量,我们处理了100个批次。为了确保可靠性,我们重复进行了每批次训练时间和吞吐量的计算50次,并将这些测量结果的分布以扩展数据图中的箱形图形式展示。10所有测试均在配备16GB显存的NVIDIA V100 GPU上进行,确保了所有实验中计算资源的一致性。
报告摘要
有关研究设计的进一步信息可在该文件中获得。自然Portfolio报道摘要与此文章相关联。
数据可用性
本研究生成的数据可从国家生物技术信息中心基因表达总论(GEO)下访问编号为GSE254493处理后的数据集可在Zenodo上获取(https://doi.org/10.5281/zenodo.10633252)70果蝇的STARR-seq数据可在GEO下通过访问号获取编号GSE183939人类MPRA数据集可从Zenodo获取(https://doi.org/10.5281/zenodo.8219231)71人类ATAC-seq数据可在GEO下通过访问号获取编号为GSE170378. 源数据本文提供了这些内容。
参考文献
菲利普斯,T. 真核生物转录和基因表达的调节。国立教育 1, 199 (2008).
罗德尔,R. G. 真核生物转录的50多年:因子和机制的扩展宇宙。自然·结构与分子生物学 26, 783–791 (2019).
克拉默,P. 基因转录的组织与调控。自然 573, 45–54 (2019).
福尔隆, E. E. M. & 莱文, M. 增强子与染色体拓扑结构的发展。科学 361, 1341–1345 (2018).
菲尔德,A. & 阿德尔曼,K. 增强子功能和转录的评估。生物化学年度评论 89, 213–234 (2020).
德博尔,C. G. & 泰帕莱,J. 拿出基因组:解决这一问题的路线图CIS监管法规。自然 625, 41–50 (2024).
泽特林格,J. 关于转录因子如何读取的七个误区cis监管法规。当前意见系统生物学 23, 22–31 (2020).
泰科等人. 高通量发现和表征人类转录效应物。细胞 183, 2020–2035 (2020).
阿拉拉索尔,N., leng,H., lin,Z.-Y., gingras,A.-C. & taipale,M. 人类细胞中转录激活因子的鉴定与功能特征分析。分子细胞生物学杂志 82, 677–695 (2022).
赖特尔,F.,魏纳罗伊特,S. & 斯塔克,A. 转录因子和辅因子的组合功能。 Current Opin Genet Dev. 43, 73–81 (2017).
阿里帕纳希,B.,德隆,A.,韦尔奥赫,M. T. & 弗雷,B. J. 利用深度学习预测DNA和RNA结合蛋白的序列特异性。自然·生物技术 33, 831–838 (2015).
周俊杰 & 罗雅斯安卡娅. 使用基于深度学习的序列模型预测非编码变异的影响。自然方法杂志 12, 931–934 (2015).
德波尔,C. G. 等。利用一亿个随机启动子解析真核基因调控逻辑。自然·生物技术 38, 56–65 (2020).
阿加瓦尔,V. & 谢恩杜里,J. 使用深度卷积神经网络直接从基因组序列预测mRNA丰度。细胞报(Cell Reports) 31, 107663 (2020).
Avsec, Ž. 等人. 通过整合长程相互作用有效预测序列中的基因表达。自然方法杂志 18, 1196–1203 (2021).
Avsec, Ž. 等人. 基于分辨率的转录因子结合模型揭示了模糊基序语法。自然·遗传学 53, 354–366 (2021).
Celaj, A. 等人。RNA 基础模型能够发现疾病机制和候选治疗药物。预印本于bioRxiv https://doi.org/10.1101/2023.09.20.558508 (2023).
Linder, J., Srivastava, D., Yuan, H., Agarwal, V. & Kelley, D. R. 从DNA序列预测RNA-seq覆盖作为基因调控统一模型。预印本在bioRxiv https://doi.org/10.1101/2023.08.30.555582 (2023).
卡普洛夫,I. M. 等人。通过预测开放染色质的组织特异性差异来推断哺乳动物的组织特异性调控保守性。BMC基因组学 23, 291 (2022).
Kelley, D. R., Snoek, J. & Rinn, J. L. Basset:使用深度卷积神经网络学习可访问基因组的调控代码。基因组研究杂志 26, 990–999 (2016).
Quang, D. & Xie, X. DanQ:一种用于量化DNA序列功能的混合卷积和循环深度神经网络。核酸研究杂志 44,e107 (2016).
瓦伊什纳夫,E. D. 等人。基因调控DNA的进化、可进化性和工程学。自然 603, 455–463 (2022).
俄塞科夫斯基,O. 等人。ImageNet 大规模视觉识别挑战。国际计算机视觉杂志 115, 211–252 (2015).
林,T.-Y. 等。Microsoft COCO:上下文中的常见对象。在第十三届欧洲会议 proceedings 计算机视觉(Fleet D., Pajdla T., Schiele B. & Tuytelaars T. 编) (Springer, 2014).
梅耶,P. & 萨伊斯-罗德里格兹,J. 系统生物学建模的进步:十年的DREAM挑战众包活动。细胞系统学 журнал Cell Systems 12, 636–653 (2021).
沙胡等. 人类基因调控元件的序列决定因素。自然·遗传学 54, 283–294 (2022).
金尼,J. B., 莫鲁甘,A., 喀兰,C. G. & 科克斯,E. C. 使用深度测序来表征转录调控序列的生物物理机制。 Proc. Natl Acad. Sci. USA 107, 9158–9163 (2010).
Sharon, E. 等人。从数千个系统设计的启动子的高通量测量中推断基因调控逻辑。自然·生物技术 30, 521–530 (2012).
夏斯特里,B. S. SNP在疾病基因定位、药物开发和进化中的应用。人类遗传学杂志 52, 871–880 (2007).
谭,M. & 黎,Q. EfficientNet:重新思考卷积神经网络的模型缩放。在第36届国际机器学习大会论文集(Chaudhuri, K. & Salakhutdinov, R. 编)(PMLR,2019)。
谭,M. & 黎,Q. EfficientNetV2:更小的模型和更快的训练。在第38届国际机器学习大会论文集(梅拉 M. & 张 T. 主编)(PMLR,2021)。
何恺明,张翔,任少卿,孙剑。深度残差学习在图像识别中的应用。在Proceedings. 2016 IEEE计算机视觉与模式识别会议(O'Conner, L. 编) (IEEE, 2016).
霍赫雷特尔,S. & 施米德胡伯,J. 长短期记忆模型。神经计算杂志 9, 1735–1780 (1997).
黄志威,徐伟,及余凯。用于序列标注的双向LSTM-CRF模型。预印本于https://doi.org/10.48550/arXiv.1508.01991 (2015).
Kingma, D. P. & Ba, J. Adam:一种随机优化方法。在国际学习表征大会(ICLR, 2015).
洛施奇洛夫,I. & 哈特尔,F. 解耦权重衰减正则化。在国际学习表征大会(ICLR,2019).
Pennington, J., Socher, R. & Manning, C. GloVe: 全局词向量的词表征。在2014年自然语言处理经验方法大会论文集(Moschitti, A., Pang, B. & Daelemans, W. 编) (计算语言学协会,2014).
姬宇,周子健,刘航及Davuluri RV. DNABERT:用于基因组DNA语言的双向Transformer预训练模型。生物信息学 37, 2112–2120 (2021).
Devlin, J., Chang, M. W., Lee, K., & Toutanova, K. BERT:深度双向变换器的语言理解预训练。在北美计算语言学协会人类语言技术分会会议 proceedings第1卷,4171-4186页(长篇和短篇论文,2019年)
德波尔,C. G. & 哈维斯,T. R. YeTFaSCo:一个评估过的酵母转录因子序列特异性的数据库。核酸研究杂志 40D169-D179 (2012).
林等。等人。亲和力优化的增强子变异干扰发育。注意,人名“Lim”在这里被译为“林”,在没有更多信息的情况下,无法进一步确定具体中文名,所以采用通用翻译方式。其余部分直接意译以符合中文表达习惯。原文作者列表中常见的"et al."表示还有其他未列出的合著者,此处保持原样不翻译。自然 626, 151–159 (2024).
阿尔梅达,B. P.,赖特尔,F.,帕加尼,M. 和斯塔克,A. DeepSTARR从DNA序列预测增强子活性并实现合成增强子的从头设计。自然·遗传学 54, 613–624 (2022).
阿诺德等人。C. D. 等人利用STARR-seq技术鉴定出全基因组范围的定量增强子活性图谱。科学 339, 1074–1077 (2013).
扎比迪,M. A. 等。增强子-核心启动子特异性区分发育基因和管家基因的调控。自然 518, 556–559 (2015).
Agarwal, V. 等人。三种不同的人类细胞类型中转录调控元件的大规模并行特征分析。预印本发表于:bioRxiv https://doi.org/10.1101/2023.03.05.531189 (2023).
潘帕里,A. 等人。染色质可及性偏置因子分解的高分辨率深度学习模型揭示了cis调控序列语法、转录因子印记和调控变异。Zenodo https://doi.org/10.5281/zenodo.7567627 (2023).
Brennan, K. J. 等人. 染色质可及性在果蝇胚胎的命运由转录因子先驱作用和增强子激活决定。开发部CELL 58, 1898–1916 (2023).
特雷维诺 AE 等人。人类发育大脑皮层的染色质和基因调控在单细胞分辨率下的动态变化。细胞 184, 5053–5069 (2021).
邓汉姆 I 等。人类基因组中的DNA元件综合百科全书。自然 489, 57–74 (2012).
Karollus, A., Mauermeier, T. & Gagneur, J. 当前基于序列的模型能够捕捉到启动子中的基因表达决定因素,但大多忽略了远端增强子。基因组生物学 24, 56 (2023).
周浩,Shrikumar A.,Kundaje A. 关于基因组学深度学习模型反向互补等变性的理解研究。在第十六届计算生物学中的机器学习会议 proceedings(Knowles, D. A., Mostafavi, S. & Lee, S.-I. 编辑) (PMLR, 2022).
庄俊等。代理间隙最小化改进了尖锐度感知训练。在国际学习表征大会(ICLR,2022).
伊扎莫夫,P.,波多普里金,D.,加里波夫,T.,维特罗夫,D.P.及威尔逊,A.G. 权重平均导致更宽的最优解和更好的泛化能力。在人工智能不确定性会议(UAI, 2018)
Dosovitskiy, A. 等。一张图片值16×16个词:大规模图像识别中的变压器。在国际学习表征大会(ICLR,2021).
刘,L.等. 关于自适应学习率方差的探讨及进一步研究。在国际学习表征大会(ICLR,2020).
林,T., 戈雅尔,P., 吉里斯克,R.B., 何,K. & 多拉尔,P. 密集目标检测的 focal loss。在IEEE国际计算机视觉大会(ICCV)2999–3007 (IEEE, 2017).
Chung, J., Gulcehre, C., Cho, K. & Bengio, Y. 关于序列模型中门控循环神经网络的实证评估。在NIPS 2014深度学习研讨会(NIPS, 2014)
Fortin, F.-A., Rainville, F.-M. D., Gardner, M.-A., Parizeau, M. & Gagné, C. DEAP:轻松使用进化算法。机器学习研究期刊(J. Mach. Learn. Res.) 13, 2171–2175 (2012).
德布尔,C. G. CRM2.0。 GitHub https://github.com/de-Boer-Lab/CRM2.0 (2023).
Langmead, B. & Salzberg, S. L. 使用Bowtie 2进行快速带缺口读取比对。自然方法杂志 9, 357–359 (2012).
德波尔,C. G.,雷,J. P.,哈科恩,N. & 雷杰夫,A. MAUDE:基于排序的CRISPR筛选中推断表达变化。基因组生物学杂志 21, 134 (2020).
Smith, L. N. & Topin, N. 超收敛:使用大学习率快速训练神经网络。在人工智能和机器学习在多域作战应用中的运用第11006卷,369-386页(SPIE,2019年)。
申샛균,欧敏英,朴young,& 金时焕。DeepFam:基于深度学习的无比对蛋白质家族建模和预测方法。生物信息学 34,i254–i262 (2018).
李玉,倪鹏,张松,李刚及苏哲。ProSampler:在大型ChIP-seq数据集中用于组合基序发现的超快且准确的基序查找工具。生物信息学 35, 4632–4639 (2019).
胡景, 沈凌, 孙刚 (2018). 压缩与激励网络。在IEEE计算机视觉与模式识别会议 proceedings7132–7141 (IEEE, 2018).
Penzar, D. 等人。LegNet:一种用于短DNA调控区域的最佳深度学习模型。生物信息学 39,btad457 (2023).
伊夫,S. & 塞格迪,C. 批量规范化:通过减少内部协变量偏移来加速深度网络训练。在国际机器学习大会(ICML, 2015)
Elfwing, S., Uchibe, E. & Doya, K. S形加权线性单元在强化学习中用于神经网络函数逼近。神经网络 107, 3–11 (2018).
Kwak, I.-Y. 等人。ProFormer:一种混合马卡龙变压器模型从启动子序列预测表达值。 BMC生物信息学 25, 81 (2024).
拉菲, A. M. 2022年随机启动子DREAM挑战赛:利用数百万个随机启动子序列预测基因表达。Zenodo https://doi.org/10.5281/zenodo.10633252 (2024).
Agarwal, V., Schubach, M., Penzar, D. & Dash, M. P. 利用大规模并行方法对三种不同的人类细胞类型中的转录调控元件进行表征。Zenodo https://doi.org/10.5281/zenodo.8219231 (2023).
致谢
我们衷心感谢来自谷歌大脑的J. Caton,他在为挑战参与者设置云资源方面提供了宝贵的技術支持。没有他的专业指导和帮助,此次挑战的成功组织是不可能实现的。我们也非常感谢TPU研究雲為所有挑戰參與者提供了必要的云端資源,確保了一個公平的競賽環境。此外,我们还感谢英属哥伦比亚大学生物医学研究中心的Underhill实验室在该项目中提供的計算資源支持,并希望他们不久后无需再升级工作站。我们还要感谢Deep Genomics为挑战中的顶尖团队提供旅行资助。这项研究部分得益于谷歌TRC、加拿大数字研究联盟和英属哥伦比亚大学高级研究计算中心的支持。本研究还得到了加拿大自然科学与工程研究理事会(RGPIN-2020-05425 至 C.G.D.)、干细胞网络(ECR-C4R1-7 至 C.G.D.)及加拿大健康研究院(PJT-180537 至 C.G.D.)的支持。C.G.D. 是英属哥伦比亚卫生研究迈克尔·史密斯学者,并且得到了美国国立卫生研究院(资助编号 K99-HG009920-01)的支持。A.M.R. 获得了英属哥伦比亚大学的4YF奖学金支持。I.V.K. 和 D.P. 得到了俄罗斯科学基金会 20-74-10075 的支持。S.K. 受到韩国政府(科学技术信息通信部)资助的信息和通信技术(ICT)规划与评估研究所赠款的支持(编号为:2021-0-01343,首尔大学人工智能研究生课程)。I.Y.K. 获得了由韩国国家研究基金会资助的科学技术信息通信部资助项目(RS-2023-00208284)的支持。
伦理声明
利益冲突
E.D.V.是Sequome公司(股份有限公司)的创始人。A.R.是Genentech公司的员工,并持有Roche公司的股权。A.R.还是Celsius Therapeutics的联合创始人和股东,以及Immunitas的股东;截至2020年7月31日,曾担任Thermo Fisher Scientific、Syros Pharmaceuticals、Neogene Therapeutics和Asimov的科学顾问委员会成员。A.R.在本研究开始时是霍华德休斯医学研究所的研究员。其余作者声明没有利益冲突。
同行评审
同行评审信息
自然生物科技感谢Žiga Avsec及其他匿名审稿人对此工作的同行评审所作出的贡献。
附加信息
出版者注施普林格·自然对于出版地图和机构 affiliation 的管辖权主张保持中立。 注:affiliation 一词在某些语境下可能没有直接的中文翻译,这里保留了原词。如果需要更详细的解释或特定术语的翻译,请告知。根据要求,只输出翻译结果且未添加任何额外信息,因此补充说明如下: 原文保持不变的情况下: Springer Nature 对于出版地图和机构隶属关系中的管辖权主张保持中立。
扩展数据
扩展数据图1 DREAM挑战公共排行榜上表现最佳团队的进步情况。
(A,B)性能(请注意缺少闭合的内容,原句似乎不完整。如果只需要翻译给出的部分,这便是其对应的中文。)_performance_(如果是要求仅基于提供的词进行翻译的话,那么结果就是:“性能”)y无实际内容需要翻译:-axes) in (APearson分数和(B参与队伍(颜色)每周取得的Spearman得分x无轴),展示了此类挑战在激励更好的机器学习模型开发方面的有效性。
扩展数据图2 自举法提供了模型预测的稳健比较。
(A,B,C)频率y等级(轴)的否定形式(无轴)x无实际内容需要翻译:-axes) in (APearson分数,(B斯皮尔曼得分和综合排名(皮尔逊得分排名与斯皮尔曼得分排名之和)对于\(n\)从测试数据集中抽取10,000个样本给表现最佳的团队。
扩展数据图4 每个测试数据子集中的团队表现。
(A,B各队模型性能(颜色和数值)y(无轴)在每个测试子集中的x无实际内容需要翻译,保持原样: -axes), for (A培生公司r2以及 (B斯皮尔曼ρ。热图颜色调板按列进行最小-最大规范化。
扩展数据图5 对SNV、基序排列和基序扰动的表达变化响应。
表情变化(y对于模体扰动最大,对于SNV最小,对于模体平铺介于两者之间。
扩展数据图6 不同可能的前三种DREAM挑战模型组合在Prix Fixe运行中的Spearman得分表现。
模块在轴上标明,顶部是数据处理器和训练器模型x轴线,底部最终层块x轴线,左侧核心层阻塞y轴,右侧为第一层区块y轴。不兼容的组合以及在训练过程中未收敛的组合已被灰色显示。
扩展数据图7 DREAM挑战赛顶级模型及其最佳对应模型的性能比较。
性能(x去掉顶部三个DREAM挑战模型的轴(如果原句意不包含被删除的内容,建议补充完整句子)y-autosome.org,BHI和UnlockDNA及其基于核心层块的最佳表现对应版本(针对不同的测试子集)。
扩展数据图9 经DREAM优化的模型的NN架构图。
(A-C)高层概述了(ADREAM-RNN, (BDREAM-CNN,和CDREAM-Attn模型。D-F核心层中使用的不同网络模块的高层次说明A-C). Vanilia卷积块、组卷积块、SE块、Stem块、FFN块、可分离卷积块和多头注意力块在详细描述中给出66并且69.
扩展数据图10 不同模型在计算效率(每批训练时间和吞吐量)和容量(参数数量)方面的比较分析。
(A, D, G)每批次训练时间(秒)y无实际内容需要翻译 原文:-axes),(开始,结束,隐藏)或其他具体含义请确认 context 信息。由于没有更多关于(E,H)的具体说明,通常情况下直接翻译或保持英文原样处理。如果必须选择一个可能的解释,则根据常见的编程或者数据标注习惯:"B" 表示边界(Begin),假设这里的"E"表示结束(End),而"H"可能是特定上下文中的代码或标签如“隐藏(Hide)”。若无额外信息,直接给出原文: (B,E,H)每秒预测吞吐量(y无轴),和(C, F, I)参数数量y无实际内容需要翻译,保持原样: -axes) for the models (x无实际内容可翻译,输出原文: -axes, colours) applied to (A-C人类ATAC-seq,D-F果蝇 STARR-seq,和 (G-I人类MPRA数据集。箱线图表示每批训练时间(A、D、G)和吞吐量(B、E、H)的测量分布,这些值重复了50次以确保可靠性。
补充信息
权利与许可
开放访问本文根据知识共享署名4.0国际许可协议发布,该协议允许任何媒体或格式下的使用、分享、改编、分发和复制,前提是您必须为原始作者及出处提供适当的引用,并链接至知识共享许可协议,并指出对内容作出的修改。如无特别说明,在文章中的图像或其他第三方材料受本文的知识共享许可协议约束,除非在相关材料的引用中另有声明。如果材料未包含在本文的知识共享许可协议范围内且您的预期用途不符合法律规定或超出了许可范围,则您需要直接从版权持有人处获得授权。要查看该许可证,请访问http://creativecommons.org/licenses/by/4.0/.
关于本文'article'在此处可能指的是文章的一个部分或者章节,如果是这样的话,可以译为“关于本节”或“关于本部分”。如果直接理解为整篇文章的介绍性说明,最准确的翻译就是“关于本文”。由于没有更多上下文信息,给出通用和直接的翻译: 关于本文

引用这篇文章
拉菲, A.M., 诺吉娜, D., 彭扎尔, D.等著しく(etal.) 注意:"etal."通常在学术引用中使用,意为“等人”,正确的翻译应为“等”或“等人”。由于提供的文本直接且简短,“等”即可准确表达原意。上述回答包含了日语的表达方式,考虑到直接翻译要求,正确的中文翻译是:“等人” 或者简单的 “等”。根据上下文习惯,通常使用“等”。 综合考虑,最简洁明了的翻译为: 等基于序列的基因调控深度学习模型的优化社区努力自然生物技术杂志(2024). https://doi.org/10.1038/s41587-024-02414-w
收到:
接受的:
发布:
DOI: https://doi.org/10.1038/s41587-024-02414-w