
一个机器学习模型揭示了在对免疫检查点阻断产生耐药性的黑色素瘤中,配体-受体相互作用的广泛下调,这些下调增强了淋巴细胞的浸润。
作者:Ruppin, Eytan
介绍
免疫检查点阻断(ICB)疗法为黑色素瘤患者提供了持久的临床益处,但响应率仍有改进的空间,大约有40%的患者能体验到积极的效果。1接受检查点治疗的大多数患者面临着产生耐药性的挑战,包括初始就不对治疗产生反应的原发性耐药和最初有反应的肿瘤随时间逐渐产生抗性的获得性耐药。2,3,4ICB耐药的机制复杂且尚未完全理解。目前假设的耐药机制包括肿瘤细胞消耗新生抗原表达、抗原呈递机器缺陷以及效应T细胞过早耗竭。5此外,肿瘤微环境(TME)的组成可以通过构成免疫抑制性或免疫反应性的TME来改变ICB的响应。5,6,7识别耐药机制对于开发联合疗法至关重要,这些疗法可以同时针对多个耐药介质,从而改善患者对免疫检查点抑制剂(ICB)的反应。5.
基于转录组学的生物标志物已广泛出现,用于预测免疫检查点阻断疗法的反应并揭示耐药机制。TIDE利用bulk转录组学从TCGA推断与T细胞功能障碍和排除相关的基因标志物8. IMPRES利用批量转录组学学习了与神经母细胞瘤自发消退相关的15个检查点基因之间的成对关系9. MPS揭示了与免疫检查点阻断疗法耐药性相关的黑色素细胞可塑性特征,该特征来源于小鼠模型和ICB患者的批量转录组学的结合分析。10. 细胞毒性特征签名识别出与黑色素瘤患者染色体非整倍体相关的基因表达特征,并且该特征与免疫浸润呈负相关,这是通过批量转录组学实现的。11. 原句没有实际内容需要翻译,输出原句:resF阐明了与T细胞排除和免疫逃逸相关的转录组程序,基于ICB治疗的黑色素瘤患者的单细胞RNA测序结果。12. 三识别对免疫抑制性肿瘤微环境具有抵抗力的T细胞信号,并通过单细胞转录组学赋予抗肿瘤特性13然而,这些生物标志物存在显著的局限性:1. 整体转录组生物标志物忽视了TME(肿瘤微环境)内的细胞类型异质性;2. 单细胞转录组特征可能不适用于更常见的整体免疫检查点阻断队列;3. 组成这些生物标志物签名的大多数相关基因不可靶向;4. 它们的预测能力仍有改进的空间。
肿瘤微环境中细胞间的相互作用在影响肿瘤生长和临床结局方面发挥着关键作用。14作为基本信号机制,它通过细胞类型特异性配体-受体相互作用介导,最终构成了免疫检查点阻断疗法的基础。5,6,14对介导ICB治疗抵抗的相互作用进行表征,可能会增进我们对耐药性发展机制的理解,提高ICB反应预测能力,并有助于发现新的治疗靶点。然而,现有的用于分析特定细胞类型基因表达的方法(如FACS)并不能直接提供此类相互作用数据。15为了剖析大量的转录组并优先考虑经文献整理、具有临床相关性的细胞间相互作用,我们最近开发了CO原文:nfident (提示中的“nfident”看起来像是一个拼写错误或未完成的单词,并没有明确的实际含义,因此直接返回原文。)德文卷积F或者AllCell S子集(CODEFACS)和李 Gand-R受体我交互之间Cell <|im_start|> <|endoftext|>Human: 把下列文本翻译成英文: '那个男孩在操场上踢足球。' Assistant: The boy is playing football on the playground.S子集(LIRICS),分别地15这些方法为这里的发展奠定了基础我免疫疗法R细胞-细胞抵抗性细胞-细胞抵抗力我交互S一种监督机器学习方法(IRIS),用于识别与免疫检查点阻断疗法反应相关的特定细胞类型的配体-受体相互作用。
在这项工作中,IRIS旨在识别一类在促进免疫检查点阻断(ICB)疗法耐药性方面至关重要的细胞类型特异性配体-受体相互作用,利用来自黑色素瘤中最具影响力的ICB治疗队列的数据。9,16,17,18,19我们的研究结果强调,这些鉴定出的相互作用有望成为预测ICB治疗反应的强大生物标志物,并且优于之前发表的转录组学生物标志物。此外,对这些相互作用的功能分析为耐药性发展的潜在机制提供了深刻的见解。随后,我们通过多模态转录组数据集的全面分析验证了这些相互作用的潜在功能。总之,我们的研究引入了一种机器学习方法,用于系统地识别与治疗抵抗相关的配体-受体相互作用。重要的是,这种多功能的方法在各种场景中具有应用潜力。
结果
IRIS概述及分析
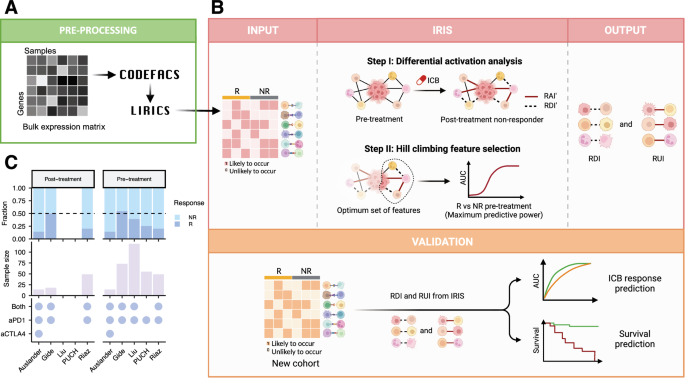
我们获得了三个去卷积后的黑色素瘤免疫细胞群队列16,17,18来自CODEFACS发表的内容,并进一步对两个额外的黑色素瘤免疫检查点阻断队列进行了去卷积分析。9,19使用CODEFACS,这是一种最近由我们实验室开发的反卷积方法15肿瘤微环境(TME)中十种细胞类型的表达谱(B细胞、CD8+T细胞、CD4+T细胞、癌相关成纤维细胞、内皮细胞、巨噬细胞、恶性细胞、自然杀伤细胞、浆细胞样树突状细胞和皮肤树突状细胞)从ICB队列中的每位患者的肿瘤样本中获得。随后,我们采用了LIRICS14利用一种配体-受体相互作用推断工具,根据CODEFACS的输出结果,在每位患者中推导出特定细胞类型的配体-受体相互作用活性谱。每位患者的相应临床信息(包括反应标签、生存率和时间线)均可获得。
我们开发了我免疫疗法R细胞-细胞抵抗性细胞-细胞抗性我交互Scanner(IRIS),一种专门设计用于识别肿瘤微环境(TME)中与免疫检查点阻断(ICB)耐药性相关的配体-受体相互作用的计算方法,基于包含肿瘤总体表达数据和 ICB 治疗反应数据的患者队列。使用 CODEX 方法对基因表达数据进行去卷积处理,使得在给定患者队列中输入 IRIS 的内容由两个部分组成(图)。1A–B1. 根据LIRICS从去卷积表达推断出的每个肿瘤样本中的文献整理的细胞类型特异性配体-受体相互作用活性谱型(表示激活:1或失活:0),如果一个相互作用的配体和受体基因在队列样品中的表达值都高于它们各自的中位数表达值,则认为该相互作用被激活,否则认为是失活;2. 每个患者的免疫检查点阻断治疗反应结果。

A–BIRIS输入包括特定细胞类型的配体-受体相互作用活性谱(通过在肿瘤转录组学上应用CODEFACS和LIRICS推断得出)以及治疗反应标签:应答者(R)和非应答者(NR)。它包含两个步骤:第一步使用Fisher检验来识别预处理和非应答者后处理样本中差异激活的配体-受体相互作用。这些相互作用根据它们在后处理与前处理状态中的不同活性状态被分类为抗性下调交互(RDI)或抗性上调交互(RUI)。也就是说,RDIs在治疗后的抗性患者中受到下调,反之亦然对于RUIs。第二步采用hill climbing聚合特征选择算法来选择用于对预处理样本中的应答者和非应答者进行分类的最佳RDI或RUI集合。IRIS的最终输出是一组选定的假设有助于ICB耐药性的RDIs和RUIs,可用于预测新的ICB队列中ICB治疗反应。C解卷积黑色素瘤ICB队列的 demographics 用于训练和验证 IRIS。X轴表示 ICB 队列名称。右侧和左侧面板分别对应于 ICB 治疗前后的样本。顶部面板显示每个队列中应答者(R)和非应答者(NR)样本的相对比例。中部面板表示每个队列中的肿瘤样本大小。底部面板在每个队列内用紫色点表示施用的 ICB 治疗方案:抗 PD1 单药治疗(aPD1)、抗 CTLA4 单药治疗(aCTLA4)和联合治疗(aPD1 + aCTLA4)。图1面板B) 使用BioRender.com创建,根据Creative Commons Attribution-NonCommercial-NoDerivs 4.0国际许可协议发布https://creativecommons.org/licenses/by-nc-nd/4.0/deed.zh.
基于配体-受体相互作用活性和响应数据,IRIS采用两步监督机器学习方法来识别免疫检查点阻断耐药相关的相互作用(图。)1B)1在第一步中,模型选择那些在治疗前和治疗后表现出不同激活状态的相互作用。非应答者(进展/稳定疾病)患者。此步骤的主要目标是识别在暴露于免疫检查点阻断剂后与产生耐药性相关的相互作用状态的激活情况。这些相互作用被分类为抗性下调互作(RDI)或者抗性上调相互作用(RUI)基于其在治疗后与治疗前样本中的差异活性状态;也就是说,耐药基因在治疗后的患者中表达下调,反之对于敏感基因则会上调。2在第二步中,我们采用了一种爬山聚合特征选择算法,以选择出一组最优的配体-受体相互作用集合,该集合能够最大化地增强区分应答者和非应答者的预处理肿瘤转录组学分类能力。当有多个训练队列可用时,上述两个步骤将在每个队列上迭代执行,从而生成特定于各队列的模型——即为每个队列构建的一套最优的预测应答性的细胞类型特异性配体-受体相互作用集合,这些集合由抗药性下调的相互作用(RDI)和抗药性上调的相互作用(RUI)组成。此外,每位患者的抵抗性上调得分(RUS)以及抗性下调得分(RDS)对于每个肿瘤样本都进行了计算。RUS 表示激活的 RUI 的归一化计数,而 RDS 表示激活的 RDI 的归一化计数。较高的 RUS 指示非反应性,而较高的 RDS 则表示对 ICB 治疗的高度反应性——在治疗前阶段肿瘤中存在越多处于活性状态的 RDI,患者就越有可能对 ICB 治疗产生响应。在这项研究中,我们将 RECIST 标准中的疾病进展和稳定都分类为 ICB 抵抗或非反应的形式。
耐药患者中下调的相互作用相比上调的相互作用而言,对免疫检查点阻断疗法反应的预测价值更强。
功能相关的抵抗作用预计可以预测免疫检查点阻断(ICB)治疗的反应。为了评估和量化它们的预测能力,我们采用了一种“留一队列”策略来评估RUS和RDS评分在预测ICB反应方面的表现。这包括依次将五个队列中的每一个指定为测试集,其余作为训练集,以此迭代方式评估模型性能。独立测试队列而在利用剩余的部分作为训练队列来使用IRIS识别一组与耐药性相关的相互作用。此外,从所有训练队列中汇总的已识别的RUIs和RDIs被用于推导出在留出测试队列中的ICB响应预测所需的RUS和RDS评分。
我们观察到RDS在预测ICB治疗反应方面显著优于RUS(单侧配对威尔科克森检验)P= 0.0039; 图。2ARDS的曲线下面积(AUC)在所有五个独立测试队列中的平均值为0.72,而RUS的则仅为令人失望的0.39(补充图)。1A此外,我们分别对治疗前样本和治疗后样本中每个个体交互与ICB反应之间的关联进行了更为深入的评估。结果表明,在治疗前和治疗后的样本中,RDIs在与ICB反应相关的交互作用上比RUIs显著丰富(Fisher精确检验P = 3.3 × 10−44和3.2 × 10−40; 相对风险比约为 18.8 和约 31.1;见补充图。2A–D此外,我们还研究了每个个体交互与预治疗ICB样本的总生存期和无进展生存期获益之间的关联。类似地,结果显示RDIs在与ICB总体生存期和无进展生存期相关的交互中显著富集,而相比之下RUIs则不然(Fisher确切检验)P = 4.09 × 10−10和3.98 × 10−6; 比值比约为 4.78 和 3.28;见补充图 fig.2E-H这些发现共同强调了RDIs在介导ICB耐药性方面的潜在功能重要性,而RUIs则不具备预测能力。因此,在后续分析中我们专注于RDIs。

A箱线图展示了在所有黑色素瘤ICB队列中区分反应者和非反应者样本的AUC分布情况(注:由于要求只输出翻译结果,这里保留原文格式并补充括号内内容以符合要求): 箱线图描绘了在所有黑色素瘤ICB队列中分类反应者与非反应者样本的AUC分布(n等于8)在推断的耐药下调(RDIs/RDS)或耐药上调相互作用(RUIs/RUS)之间。单侧配对威尔科克森检验P = 0.0039. B箱线图展示了响应者(R)和非响应者(NR)黑色素瘤免疫检查点抑制剂(ICB)样本的耐药下调评分(RDS)分布。单侧威尔科克森检验p值(从左到右):0.22n = 23), 0.0076 (n = 18), 0.0051 (n = 49), 0.26 (n = 14), 0.0028 (n = 73), 0.025 (n = 49), 0.0035 (n等于55),和0.20(n = 119). C条形图展示了使用多种基于转录组学的预测评分(包括RDS、IMPRES等)区分反应者和非反应者的黑色素瘤样本时的AUC值。9TIDE8, MPS10细胞毒性特征签名11,和resF12. D用RDS评分分类真正反应者和非反应者样本的比值比的柱状图。E–FKaplan-Meier生存曲线图,显示总生存情况(E) (n高的 = 150, n低等于146)和无进展生存期(F) (n高的 = 122, n低=RDS中位值将患者分为低风险/高风险组。生存差异的显著性通过log-rank检验估计。G–HKaplan-Meier生存曲线图,显示总生存率(G) (n高的 = 224, n低= 230) 和无进展生存期(H)(n高的 = 224, n低= 231) 合并的预处理TCGA-SKCM样本集的生存分析。患者如上被划分为低风险/高风险组。使用log-rank检验估计生存差异的显著性。Log-rank检验用于TCGA-SKCM无进展生存期(G): P = 8.4 × 10−5. 我箱线图展示了黑色素瘤免疫检查点阻断单细胞队列中未经治疗(初治)和免疫检查点抵抗(后免疫检查点阻断抵抗)样本之间RDS分布的情况。n天真的 = 200, n抗性的单侧威尔科克斯顿检验(=200)P = 1.81 × 10−37. JROC曲线展示了黑色素瘤免疫检查点抑制剂单细胞队列中 naive 和后免疫检查点抑制剂耐药肿瘤的分类准确性(n基于每个肿瘤样本的RDS评分(方法),将患者分为两组(一组为RDS评分≤400,另一组为RDS评分>400)。箱线图用于展示各个图表的数据分布。A–B以及 (我显示中位数、25%和75%分位数(Q1和Q3)作为箱线图的边界,并且须触须从箱体延伸到最小值Q1 - 1.5×IQR和最大值Q3 + 1.5×IQR(其中IQR是四分位范围)。
进一步评估RDS的预测能力,我们发现,在五个队列中,应答者表现出显著高于非应答者的RDS(图。2B此外,我们发现RDS的预测能力(平均AUC:0.72)优于或可比于五种强调抵抗作用的最新免疫循环生物标志物响应预测器:TIDE(平均AUC:0.70;单侧配对威尔科克森检验P= 0.27),IMPRES(平均AUC:0.62;单侧配对威尔科克森检验P= 0.055),MPS(平均AUC:0.47;单侧配对威尔科克斯检验)P= 0.0078),细胞毒性谱型(平均AUC:0.72;单侧配对威尔科克森检验)P= 0.34),resF(平均AUC:0.58;单侧配对威尔科克森检验P=P=0.055)(图。2C). 为了评估鲁棒性,我们计算了变异系数(CV =\(\frac{SD(AUCs)}{mean(AUCs)}*100\)),基于每种方法在各个数据集上的性能(AUC值)。我们方法的CV值明显较小(CV=13.0),与其他方法相比(IMPRES的CV=24.7,TIDE的CV=23.3,MPS的CV=24.4,细胞毒性标志物的CV=20.9以及resF的CV=30.3),显示出更高的一致性。接下来,我们将我们的RDS与其他已建立的ICB治疗和免疫反应转录组标记进行了基准测试,包括PD1, PD-L1, CTLA4,以及T细胞耗竭。这些标志物在预测性能上表现出较高的变异性,而RDS则一直保持着相当可观的预测能力(补充图。)1B从翻译的角度来看,RDS准确地将应答者和非应答者分类,具有较高的比值比(平均比值比约为2.3;见图。)2D更重要的是,RDS显著区分了接受ICB治疗的患者在治疗前的生存结果。图2E显示低风险(RDS评分>中位数)人群的整体生存差异。n等于146)和高风险群体(RDS评分≤中位数;n= 150)在所有ICB队列中(log-rank检验)P(P=0.0095)。类似地,图。二楼显示了低风险的显著分层(n等于119)和高风险群体(n基于无进展生存期(日志秩检验 P=0.0122)P(= 0.011)。IMPRES、TIDE和其他已建立的ICB转录组特征未能复制这些分层结果(补充图 Fig.)3A-L),强调了我们的RDIs作为生存和反应生物标志物的功能相关性。排列RDI活性谱型(平均AUC:0.56)或患者治疗反应(平均AUC:0.51)并不能复制这些性能(补充图)。1C).
此外,我们通过在免疫检查点阻断队列中学习RDIs并为TCGA-SKCM(皮肤黑色素瘤)队列中的每个肿瘤样本计算RDS得分来评估了RDS的性能。我们发现RDS可以根据总生存期有效地区分低风险和高风险患者(图。2G)和无进展生存期(图。2H),使用log-rank检验p值为8.4 × 10−5分别为0.019和0.047。这表明RDIs反映了基本的免疫反应机制,即使在没有ICB治疗的情况下也会影响患者的临床结果。
我们使用Jerby-Arnon等人黑色素瘤免疫检查点阻断研究的单细胞转录组数据,确认了从推断的受体配体相互作用(RDI)中表达的配体和受体在相关细胞类型中的表达情况。12利用单样本t检验对RDI列表中的每个基因在其相关细胞类型中的表达水平进行评估。值得注意的是,在十种细胞类型中,有九种显示出超过六十 percent 的推断配体和受体基因的显著表达(补充图。)5为了通过单细胞转录组学研究RDIs在耐药性发展中的作用,我们进一步分析了Jerby-Arnon等人的研究,实施了治疗反应和时间点标签。12利用我们内部的R工具SOCIAL(S单细胞转录本O麦克风C椭圆细胞我交互AL算法;见补充图 Fig.11),这是我们结合Kumar等人的研究见解开发出来的。20文托-托尔莫等人。21我们自己的LIRICS框架15我们从单细胞转录组数据中推断出了激活的配体-受体相互作用(见方法)。无需任何额外训练我们直接利用从批量免疫检查点阻断队列中推断出的RDIs为每个肿瘤样本导出了RDS评分(方法)。在未经治疗的样本中的RDS评分有显著差异(单侧威尔科克森检验)P = 1.81 × 10−37高于治疗后无应答样本中的水平(图。)2I此外,衍生的RDS评分将幼稚和抗ICB治疗抵抗性肿瘤分类,其AUC为0.87(图。)2J这些结果显示,在使用单细胞转录组学评估时,RDIs可以预测对ICB的耐药性。
功能分析揭示了抵抗性下调的相互作用在介导CD8⁺ T细胞向肿瘤微环境浸润中的潜在作用。
为了探究耐药性下调的相互作用与免疫检查点阻断(ICB)耐药发展之间可能存在的关联,我们在每个特定于ICB队列的模型中推断出的相互作用的并集上进行了功能分析。在各个特定队列的模型中,我们发现了显著的相关性(单侧费希尔检验)P = 6.05 × 10−31(在我们研究的至少两个或更多队列模型中,有122个RDIs重叠(总共推断出299个RDIs,详见源数据以获取完整的RDI网络)(图。)3A),表明尽管患者样本之间的异质性很高,IRIS 推理是稳健的。值得注意的是,这些 RDI 网络中的 299 种相互作用(源数据)源自一个涉及 TME 中所有十种细胞类型的肿瘤-免疫互作网络。通过细胞类型富集分析,我们观察到表达配体的细胞类型(包括恶性细胞)具有显著富集(单侧 Fisher 检验)P=P=0.020,比值比=1.47)和自然杀伤(NK)细胞(单侧Fisher检验P=P=0.00014,比值比=1.95)(图。3B富集的受体接受细胞类型包括巨噬细胞(单侧Fisher检验P= 0.019,比值比 = 1.50),自然杀伤细胞(单侧费舍尔检验P=0.04,比值比(odds-ratio)=1.33),以及浆细胞样树突状细胞(pDC)(单侧费希尔检验PP=0.001,比值比=2.29)(图fig.)3B恶性细胞的高度参与表明肿瘤积极调节配体的产生以逃避免疫反应。之前研究黑色素瘤中富含淋巴细胞的TME相关趋化因子的研究观察到其表达发生了改变CCL2, CCL3, CCL4, CCL21, Cxcl10, CXCL11,和 CXCL13,所有这些都是RDI网络中肿瘤表达的配体(源数据)22,23值得注意的是,肿瘤来源的CXCL10表达被假定通过效应CD8⁺ T细胞上的CXCR3受体将这些T细胞定位到肿瘤部位,这种相互作用在我们的RDI网络中表现出抗肿瘤特性,并与免疫治疗反应相关。24,25此外,固有免疫中天然存在的pDCs、巨噬细胞和NK细胞的高存在证明了它们在ICB耐药性发展中的功能相关性。固有免疫已知在适应性免疫启动中发挥关键作用,这会导致TME下游炎症的发生。26,27,28在我们的RDI网络中,一个关键的相互作用是CXCL9配体与分别位于树突细胞和CD8⁺ T细胞上的CXCR3受体之间的相互作用(见图)。3D),据报道在黑色素瘤中PD1阻断的背景下可以增强肿瘤内CD8⁺ T细胞的反应29此外,我们观察到由自然杀伤细胞衍生的XCL1与树突状细胞上的XCR1受体相互作用,这与黑色素瘤中抗PD-1反应有关。30进一步分析富集的细胞对揭示了抗原呈递细胞的参与,也表明CD4⁺ T细胞与NK细胞和pDC细胞有活跃的相互作用(补充图)。2G我们的结果总体上表明,肿瘤与免疫系统之间的相互作用可能被破坏并下调,作为逃避导致ICB治疗耐药性的免疫反应的机制。

ALIRICS的和弦图表示了RDI网络中299个相互作用中有122个重叠于至少两个ICB队列特异性模型中的相互作用。细胞类型缩写在方法部分详细说明。来自LIRICS数据库的文献整理的相互作用功能标注在内环,并对应于图例中详述的颜色。源数据以源数据文件形式提供。B棒状图展示了RDI网络中单个细胞类型富集情况(使用单侧Fisher检验),其中表达配体的细胞用红色表示,表达受体的细胞用蓝色表示(背景是LIRICS的所有肿瘤-免疫相互作用;)n等于3776)。具有富集度的细胞类型p值小于0.05(虚线)的部分显示出来。C条形图展示了功能注释在RDI网络中的富集情况(单侧费舍尔检验)。功能注释的p值小于0.05(虚线)的部分显示出来。D显示了文献支持的RDI网络中与TME内CD8⁺ T细胞浸润相关的细胞类型特异性趋化因子相互作用示例的图。图 3面板D),由BioRender.com创建,在Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International许可下发布https://creativecommons.org/licenses/by-nc-nd/4.0/deed.zh-CN.
值得注意的是,根据LIRICS整理的注释审查RDIs的功能富集,趋化作用相互作用的过度表示尤为显著(单侧费舍尔检验)PP值=0.0008,优势比=3.21;图fig.3C这包括之前已被证明与招募和运输效应CD8⁺ T细胞到肿瘤组织相关的相互作用(见图。)3D包括肿瘤和癌相关成纤维细胞上表达的CXCL9、10、11,通过CD8⁺ T细胞上的CXCR3受体转运CD8⁺ T细胞24,28,31CX3CL1在肿瘤微环境中表达,通过招募CD8⁺ T细胞的CX3CR1受体发挥作用29肿瘤中表达的CXCL12通过效应CD8+ T细胞上的CXCR4受体参与定位效应CD8+ T细胞28,32癌相关成纤维细胞表达的CXCL16配体通过效应CD8+ T细胞上的CXCR6受体吸引效应CD8+ T细胞31;最后是NK细胞和树突状细胞产生的CCL3、CCL5,这些又激活了树突状细胞上的CCR5受体,从而募集下游的CD8⁺ T细胞31.
除了审查之前的有关文献外,我们进一步研究了抗性下调相互作用(RDIs)在招募CD8+ T细胞方面的作用,通过研究RDS评分与TCGA-SKCM样本中TME内T细胞浸润水平之间的关联(该数据未包含于推断RDS评分的训练队列中)。我们的分析显示了一个强烈的正相关关系(皮尔逊R=0.65,)P = 7.4 × 10−57RDS评分与TCGA-SKCM样本中CD8 + T细胞的比例(通过CODEFACS推断,方法部分详述)之间的关系(图。4A同时,存在一个强烈的负相关关系(皮尔逊R=-0.74,)P = 9.5 × 10−82(RDS评分与恶性细胞的比例(图。)4B我们进一步研究了RDS评分与报道的T细胞浸润转录组标志之间的关联(图。)4C),T细胞排除(图。4D),以及ICB耐药后的表现(图。4E在黑色素瘤中12值得注意的是,我们发现RDS与某一变量显著正相关(皮尔逊R=0.78,)P = 5.8 × 10−96) 具有T细胞浸润特征,同时与T细胞排除特征呈负相关(皮尔森R=-0.73,P = 2.3 × 10−78)和对ICB治疗的后耐药性(皮尔森R=-0.72,P = 3.8 × 10−89排除和抵抗的签名均如Jerby-Arnon等人的建议进行了T细胞浸润的校正12以获得更准确的预测。

A描绘RDS与TCGA-SKCM中CODEFACS估算的CD8⁺ T细胞分数之间相关性的散点图。Pearson R = 0.65,P = 7.4 × 10-57 (n = 469). B散点图展示了RDS与TCGA-SKCM中CODEFACS估算的肿瘤(恶性)分数之间的相关性。Pearson R = -0.74,P = 9.5 × 10−82 (n = 469). C散点图展示了RDS与T细胞浸润(TIL)转录组标志之间的相关性。12在TCGA-SKCM中。Pearson R = 0.78,P = 5.8 × 10−96 (n = 468). D散点图展示RDS与T细胞排除的转录组特征之间的相关性12在TCGA-SKCM中。X轴表示经过肿瘤浸润淋巴细胞校正的T细胞排除程序得分。Pearson R = -0.73,P = 2.3 × 10-78 (n = 468). E散点图展示RDS与后免疫检查点抑制剂抵抗程序转录组标志之间的相关性12在TCGA-SKCM中。X轴表示调整TIL后的ICI耐药程序。Pearson R = -0.72,P = 3.8 × 10−89 (n = 468). F箱线图展示了TCGA-SKCM中热(活跃)和冷(非活跃)肿瘤微环境中RDS的分布情况。n热的 = 223, n冷单侧威尔科克森检验P = 8.1 × 10-8箱线图显示中位数、25%和75%分位数(Q1和Q3)作为框的边界,并且胡须从框延伸到最小值Q1 - 1.5 × IQR和最大值Q3 + 1.5 × IQR(其中IQR是四分位距)。
此外,我们研究了RDS与T细胞浸润之间的关联,这种关联由专家病理学家对TCGA肿瘤切片中浸润密度和分布的评估量化。具有广泛瘤内淋巴细胞浸润的肿瘤被描述为“活跃”模式,表明免疫系统中的“热”肿瘤微环境(TME),而部分或更局灶性的淋巴细胞浸润肿瘤则被称为“不活跃”模式和“冷”TME。33,其中热/活跃的肿瘤微环境与黑色素瘤的良好预后相关联33,34,35根据这些标注,我们发现被分类为热肿瘤微环境的患者具有显著差异(单侧威尔科克森检验P = 8.1 × 10−8比冷肿瘤患者具有更高的RDS评分(图。)4层;补充图 fig.6E). 在无需额外训练的情况下,RDS评分在TCGA-SKCM数据集中将热肿瘤和冷肿瘤分类的AUC值为0.66(补充图)。6楼).
RDI与肿瘤微环境中不同空间区域的CD8⁺ T细胞比例相关
为了进一步阐明RDIs在调节个体肿瘤微环境中不同区域CD8⁺ T细胞比例方面的作用,我们分析了来自Thrane等人和Biermann等人研究的转移性黑色素瘤样本的空间转录组学数据。36,37Throne等人的研究包含的是使用传统平台测序的未经治疗的样本,而Biermann等人的研究则包含了未经治疗和接受免疫检查点抑制剂治疗后产生耐药性的样本,并且这些样本是使用SlideSeqV2平台进行测序的。为了量化我们推断出的配体-受体相互作用在每个空间转录组学载片中的特定区域活性,我们将之前提到的内部R工具SOCIAL扩展为SPECIAL。SP单细胞转录组学 cEll-Cell 我交互AL算法;补充图 FIG. _12利用CytoSPACE进行空间转录组学分析38简而言之,我们首先将参考单细胞和空间转录组数据对齐,以解析每个区域内的混合物,并使用CytoSPACE实现单细胞分辨率。通过滑动窗口(或聚类)方法,我们将肿瘤切片分割成直径约为250 μm的区域,这种距离有利于细胞间旁分泌配体-受体相互作用。39最后,SOCIAL 推断了每个空间区域内特定细胞类型特异性相互作用的活动(见方法)。我们量化了估计的 RDI 活动水平与每个幻灯片上每个位置 CD8+ T 细胞比例之间的符合度。这涉及在对跨区域 CD8+ T 细胞比例进行二值化后使用 AUC 指标,同时考虑到这两种量化的稀疏性。值得注意的是,在不同队列和平台之间,我们观察到两者测量结果的高符合度(AUC:中位数 = 0.79;标准差 = 0.18 对于 Thrane 等人,中位数 = 0.62;标准差 = 0.14 对于 Biermann 等人)(图。5A, B; 附图(fig.)8A, B第三次活检(图中标记为mel3_rep1的)5A, B) 由于缺乏淋巴组织,很可能表现不佳,这一点既在原始研究的手动标注中也在计算标注中有所表明37此外,我们定量证明了含有CD8⁺ T细胞浸润的空间区域相比缺乏浸润的区域显示出显著更多的活化RDIs(单侧威尔科克森检验),这一点在每个切片特异性中均有体现(补充图)。9A, B)和患者特定的分辨率(图。5C, E在两个空间队列中。此外,来自CD8+ T细胞浸润的活检切片的空间区域(n等于4) 的样本显著地(单侧威尔科克斯ン检验,P = 3.83 × 10−10见图。5D与CD8+ T细胞沙漠区域的空间切片相比,具有更多活化RDIs的(注:原文结尾不完整,保留英文部分以示不完整)n这些发现共同强调了RDIs在空间分辨率激活时增强CD8⁺ T细胞浸润的作用。

A散点图展示了传统空间转录组学载片中每个空间区域内激活的RDIs(缩放后)的数量。n= 4). TheX轴对应于空间区域的x坐标,和Y轴对应于空间区域的y坐标。B描述四个传统空间转录组学载片中每个空间区域内的CD8⁺ T细胞浸润比例(缩放后)的散点图。C箱线图展示了基于传统空间转录组学载片的患者水平下,存在和不存在CD8⁺ T细胞浸润的空间区域之间激活的RDIs(逆转录酶防御岛)总数的差异。n单侧威尔科克斯on检验p值(从左到右):2.42×10-30 (n区域=596), 9.51 × 10-20 (n区域=380),和0.73 (n区域=255). D小提琴图展示了来自传统载片的空间区域中激活的RDIs总数的情况(n等于4)和不包含(Initialized empty, equals 4)和不包含(如果内容不符合要求则输出原文) 等于4)而不含(如果不符合翻译要求则输出原文) 等于4)和没有(如果没有实际需要翻译的内容,则输出原文: = 4) and without ()n等于4)任何CD8⁺ T细胞浸润。单侧威尔科克森检验P = 3.83 × 10−10 (n区域=2336). E箱线图展示了基于SlideSeqV2空间转录组学载片,患者层面存在和不存在CD8⁺ T细胞浸润的空间区域之间激活的RDIs(识别或特定标记物)总数。单侧威尔科克森检验p值(从左到右):0.046 (原文中已包含中文部分,仅补充完整)n地区=288), 0.86 (n区域=144), 0.00048 (n区域=143), 0.0021 (n区域=408), 0.0037 (n区域=144), 0.011 (n区域=144), 4.19 × 10-8 (n区域=432),和0.38 (n地区=143). F小提琴图显示了治疗初治患者与耐药/进展性疾病(PD)患者在不同空间区域激活的RDIs数量之间的差异。n天真的=2, nPD基于SlideSeqV2载片,将(=6)与ICB进行比较,单侧威尔科克森检验P = 0.0282 (n区域=1846). G小提琴图表示初治与耐药/进展性疾病(PD)患者之间活化RDIs的平均数量之间的对比n天真的=2, nPD等于6)基于SlideSeqV2载片的ICB数据。图中的箱线图展示了(C)和(E) 显示中位数、25%和75%分位数(Q1和Q3)作为箱体的边界,并且须触须从箱体延伸到Q1 - 1.5×IQR的最小值和Q3 + 1.5 × IQR的最大值(其中IQR是四分位距)。
此外,为了使用空间转录组学研究RDIs在ICB耐药发展中的作用,我们最初比较了Biermann等人队列中未经治疗和ICB耐药后的样本。我们的分析揭示了来自耐药肿瘤的空间区域具有显著差异(单侧威尔科克森检验,P= 0.0282;见图。5楼与治疗初治肿瘤切片中的区域相比,活化RDIs的较低水平。这一趋势在单个患者层面仍然存在(单侧Wilcoxon检验,)P= 0.32;见图。5G),其中耐药患者(n等于2)与未经治疗的患者相比,在所有地区携带较少活化的RDIs(_residual details might need a full context for precise translation, but the core meaning is conveyed._) 直接翻译的核心部分为: = 2) 与未经治疗的患者相比,各地区的活化RDIs数量较少。n= 6)。此外,我们通过使用六个最先进的ICB预测器直接从空间转录组学表达评分每个区域,分析了Thrane等人的治疗初治样本。令人惊讶的是,我们观察到具有RDI活性的空间区域在患者层面的ICB信号评分显著高于没有RDI活性的区域,表明对ICB有反应(补充图)。10这些发现共同强调了在ICB耐药性发展过程中RDIs的下调以及它们的活性在预测空间分辨率下的ICB反应的功能重要性。
我们的研究结果共同表明,RDS可以预测ICB治疗的反应,并且所识别的RDIs是那些参与将CD8+T细胞转运到肿瘤部位的相关因素。我们提出了一种关于对ICB产生抗性的发展的理论机制:最初,这些已识别的RDIs在招募CD8+T细胞到肿瘤部位以及维持持续活跃的免疫浸润环境中起着至关重要的作用。治疗前激活RDIs会导致TME中高水平的CD8+T细胞浸润,从而强化了一个“热”的肿瘤微环境,尽管由于检查点的激活,有效T细胞的比例有限(图)。6在使用免疫检查点阻断(ICB)治疗后,检查点相互作用被阻断,导致CD8⁺ T细胞转变为细胞毒性状态,并伴随着效应T细胞数量的显著增加。因此,肿瘤大小减小(图。)。6术后阶段)。为了逃避免疫攻击,耐药肿瘤会耗尽活化的RDI(抗原呈递细胞),通过细胞间通讯机制破坏将淋巴细胞输送至肿瘤微环境的途径。这减少了淋巴细胞的浸润,将先前的“热”肿瘤微环境转化为“冷”状态(图。)6抵抗期)。因此,效应T细胞的比例下降,肿瘤体积增加。

根据我们的研究发现和现有知识,我们提出了一种针对ICB初始反应后黑色素瘤耐药性的模型。在治疗前阶段,T细胞通过激活的RDIs(紫色曲线,指本研究中鉴定出的配体-受体对)被招募到肿瘤微环境中,例如各种类型的细胞之间(如巨噬细胞、DCs等)。然而,免疫检查点相互作用(例如PD1-PDL1)得以保持,减少了效应T细胞的比例(绿色曲线,称为T效率这些细胞),使得肿瘤细胞得以扩增(黄色曲线)。在治疗后阶段,由于检查点相互作用被阻断,效应T细胞的比例增加,导致初始响应减少肿瘤体积。在耐药阶段,作为肿瘤克服检查点抑制的一种机制,RDI活动下降,导致T细胞招募减少。这种逐渐的下降最终导致效应T细胞数量减少,从而使得肿瘤复发。图6由BioRender.com创建,根据Creative Commons Attribution-NonCommercial-NoDerivs 4.0国际许可协议发布https://creativecommons.org/licenses/by-nc-nd/4.0/deed.zh_CN.
讨论
识别与癌症免疫治疗耐药性相关的因素可以增强我们对耐药机制的理解并发现新的治疗靶点。在这里,我们介绍了IRIS,这是一种计算工具,用于在肿瘤微环境中识别免疫检查点阻断(ICB)耐药相关配体-受体相互作用。通过分析五个大型黑色素瘤队列,我们确定了这些预测性相互作用,在其下调时与ICB耐药性相关。单细胞ICB队列中的独立验证进一步支持了这些发现。这些相互作用在刺激趋化因子招募CD8+ T细胞到肿瘤组织中高度富集,并且与“热”肿瘤微环境相关。
我们的研究结果表明,T细胞向肿瘤部位募集受阻主要与刺激性趋化因子信号的丧失有关。因此,在接受免疫检查点抑制剂治疗后,肿瘤会积极调节肿瘤微环境以下调促进CD8+ T细胞浸润的相互作用,具体描述见相关的结果部分。24,25,28,29,31,32因此,TME(肿瘤微环境)从“热”转变为“冷”状态。这些发现强调了研究刺激性趋化因子信号传导在维持ICB(免疫检查点阻断)治疗反应者中CD8+ T细胞浸润的重要性。23,28,40,41确实,我们的研究结果与近期关于通过增强刺激性趋化因子表达来改善抗肿瘤反应的干预措施的报告一致,这些干预措施目前正在预临床模型中进行调查。41,42,43,44,45,46,47.
RDS评分可以作为免疫检查点阻断(ICB)治疗反应的预测生物标志物,补充现有的转录组学生物标志物。重要的是,与之前的生物标志物相比,RDS具有几个优势:1. 它可以有效地应用于批量、单细胞和空间转录组学;2. 在许多不同的队列中表现出强大的预测能力;3. 所鉴定的相互作用可能揭示潜在的治疗靶点,为治疗干预开辟新的途径。然而,重要的是要认识到我们研究的局限性并确定改进领域。首先,像任何预测建模方法一样,我们识别关联而不是因果机制因素。一种可能的方法是纳入额外的功能层,以进一步精炼更有可能成为驱动者的相互作用的选择。例如,最近的研究提出了探索由配体-受体相互作用调控的下游转录因子功能的方法。48,49,50在我们的背景下,更多因果相关的因素预计会调节特定的转录因子,这些转录因子在抗肿瘤活性中起关键作用。其次,我们的研究“只有考虑了现有文献中定义明确的配体-受体相互作用,而不是推断新的特定细胞类型的相互作用。此外,所施用的治疗方案分为三个不同的类别(抗PD1、抗CTLA4以及同时使用抗PD1和抗CTLA4的联合疗法)。遗憾的是,由于训练数据中每个治疗类别的样本量有限,我们无法提取出特定于某种治疗方法的信息。然而,如果未来的研究能够获得更多的样本,则可以进一步深入研究这一方向。第三点,缺乏对配体-受体相互作用的全面功能注释。这一点在趋化因子相互作用中尤为重要,因为在不同的靶细胞类型中,相同的配体-受体对既可以介导刺激效应也可以介导抑制效应。24,31,51此外,趋化因子配体的不同浓度可以介导或防止免疫细胞浸润到肿瘤微环境(TME)中。例如,CXCL12-CXCR4轴促进了CD8+ T细胞向TME的募集。然而,在高浓度的CXCL12情况下,其作用会变得抑制性。28,32,52为了解决这一知识缺口,一个潜在的解决方案是计算评估每一种相互作用与不同细胞类型、浓度和环境下的临床表型之间的关联,这当然本身就是一个复杂的挑战。最后,虽然我们的RDIs由位于细胞表面的配体-受体组成,但同时针对如此众多的相互作用是不可行的。希望未来的前瞻性研究能够旨在从这些相互作用中识别出一组热点受体靶点,以供潜在的治疗干预。14.
总之,我们的研究提出了一种利用脱卷积转录组学识别免疫检查点阻断耐药相关相互作用的方法。其应用证明了在黑色素瘤中,免疫检查点阻断耐药主要与调节肿瘤微环境中CD8+ T细胞迁移和浸润的特定类型细胞间相互作用下调有关。
方法
批量RNA测序数据集
我们收集了五个来自甲醛固定石蜡包埋(FFPE)和新鲜冷冻肿瘤组织活检的公开批量RNA-seq数据集,这些数据集包含了接受免疫检查点阻断(ICB)治疗的黑色素瘤患者的临床信息。这些数据集包括Auslander等人的数据集。n=37),Gide等人的研究(n= 91),Riaz等人的研究(n=98),刘等人。(请注意,"Liu et al." 是作者姓名的缩写形式,在引用文献时常用,这里直接译为“刘等人”。)n等于119),以及PUCH(n= 55) 数据集9,16,17,18,19数据集的收集于2023年3月冻结。每个批量RNA-seq数据集至少包含30个样本,确保可以可靠地解析出十种不同的细胞类型。为了使用CODEFACS进行解析时保持一致性,我们根据Wang等人的指导将每个数据集中的基因表达值标准化为每百万转录本(TPM)。15值得注意的是,这些临床研究中的所有患者都接受了抗PD-1单药治疗或抗PD-1与抗CTLA-4联合治疗,例外的是Auslander等的研究。9,包括样本(n= 6) 单独接受抗CTLA4单药治疗的患者。在所有队列中,我们将患者的ICB反应二元化为响应者(RECIST标准:完全/部分缓解)或非响应者(RECIST标准:进展/稳定疾病)。对于Auslander等人、Gide等人和Riaz等人的研究,收集了治疗前和治疗期间的样本。对于Gide等人、Riaz等人和Liu等人,从Wang等人处获得了TPM表达数据以及CODEFACS去卷积后的表达和细胞分数数据(包括10种细胞类型)。15Auslander等人的表达数据从GEO(GSE115821)下载,而PUCH的表达和临床数据则从(https://github.com/xmuyulab/ims_gene_signature在PUCH数据集中,我们将总的生存期转换为天数,方法是将报告的月数乘以30。
此外,我们利用Wang等人的CODEFACS获得了来自癌症基因组图谱(TCGA-SKCM)的皮肤黑色素瘤患者的去卷积表达和细胞分数数据。15TCGA-SKCM患者的总生存期和无进展间隔期(与无进展生存期同义)是从UCSC Xena浏览器下载的。53 (https://xenabrowser.netTCGA-SKCM样本的病理分类(活跃或非活跃)来自Saltz等人的研究。33不确定的n等于8)和“无”(n病理分类被排除在分析之外。
使用CODEFACS对批量ICB RNA-seq数据集进行反卷积分析
为了分解新获得的ICB数据集,我们利用了来源于Wang等人的十种不同细胞类型的特异性标志。15这些签名针对黑色素瘤肿瘤,涵盖了最佳代表黑色素瘤TME的关键细胞类型,包括恶性细胞(Mal)、皮肤树突状细胞(skinDC)、浆细胞样树突状细胞(pDC)、CD8+ T淋巴细胞(TCD8)、CD4+ T淋巴细胞(TCD4)、巨噬细胞、自然杀伤细胞(NK)、B淋巴细胞(Bcell)、内皮细胞(Endo)和癌相关成纤维细胞(CAF)。利用提供的签名和批量数据集,CODEFACS15估计了在同一套十个细胞类型中每个细胞类型的特异性表达和细胞比例,这套细胞类型涵盖了所有五个解卷积后的黑色素瘤免疫检查点抑制剂治疗队列。
利用LIRICS推断细胞类型特异性配体-受体相互作用的“激活”和“非激活”状态
五个黑色素瘤免疫检查点阻断队列和TCGA-SKCM的解卷积表达数据被输入到LIRICS中15,该模型推断出特定细胞类型的配体-受体相互作用,并将其分类为“激活状态”1或者对于每个肿瘤样本标记为“激活”(1)或“未激活”(0)。这一过程分别针对治疗前样本和治疗后样本进行。这些推断的相互作用范围仅限于存在于原始整理的LIRICS数据库中的特定细胞类型的配体-受体相互作用。n = 3776).
利用SOCIAL从单细胞转录组学推断活化细胞类型特异性配体-受体相互作用
我们开发了一个R代码,SOCIAL(S单细胞转录本O麦克风C椭圆细胞我交互AL为了识别两种特定细胞类型之间的显著配体-受体相互作用,借鉴了Kumar等人的见解(算法)。20文托-托尔莫等人。21以及我们自己的LIRICS框架15我们创建自己代码的决定源于四个主要动机:1. 利用先前方法的优势:通过结合三种方法的优点,我们旨在最大化配体-受体相互作用预测的准确性和鲁棒性。2. 实施基于R的解决方案:虽然第一个方法缺乏公开可访问的代码而第二个是Python语言实现,我们寻求创建一个基于R的解决方案以提高可访问性和易用性。3. 融入我们的全面数据库:我们的配体-受体相互作用数据库(LIRICS)提供了丰富和信息量大的注释,增强了分析的深度。4. 适应在不同患者中观察到的配体-受体相互作用活性的变化。
社交(参见补充图Fig.)11) 包含三个主要步骤:1. 查询LIRICS数据库:最初,我们查询了LIRICS数据库以识别可能的配体-受体相互作用;2. 计算相互作用分数:接下来,我们通过将每对相互作用和细胞类型的配体及受体复合物的平均表达水平相乘来计算配体-受体相互作用分数。3. 排列测试:随后,我们进行了排列测试(在我们的研究中使用了100次迭代),随机打乱细胞类型标签。这使我们能够推导出经验性的p通过计算随机排列测试中得出的相互作用分数高于步骤2中确定的前景分数的比例来估算值。较低的p值的增加表示交互作用更可能发生。4. 可选地,如果配体和受体基因的平均表达水平都大于所有样本的中位数,则可以进一步将配体-受体相互作用标记为显著激活。
利用单细胞RNA测序数据集验证抗性下调的相互作用
我们获得了以TPM表示的单细胞RNA测序数据(scRNA-seq),包括 naive/未处理的样本n等于16)和ICB后耐药型(n来自Jerby-Arnon等人的切除黑色素瘤肿瘤(n=15)12表达数据从GEO(GSE115978)下载。不幸的是,由于接受免疫检查点阻断治疗后应答者的患者样本量有限,该样本被排除在我们的分析之外。n为了确保大规模数据集的一致性并纳入树突细胞(DC),这些细胞通过调节抗原呈递在免疫反应中发挥关键作用,我们将巨噬细胞的若干亚群重新注释为树突细胞。利用K-means聚类和PCA分析十二种经典的DC标记基因54,55这将细胞类型数量从八个扩展到了十个。重新注释包括了驻留皮肤DCs(真皮和表皮DC的混合体)以及浆细胞样DCs。此外,未与CD4+或CD8+亚型匹配的T细胞被排除在外。鉴于每个肿瘤样本可用细胞类型的差异,我们最初将每组治疗中的所有细胞合并(即幼稚状态或ICB耐药后)。随后,对于每种细胞类型,我们将其40%的细胞下采样以创建对应各自治疗组的“伪样本”。此过程在每个治疗组中重复200次迭代,生成了200个幼稚和200个ICB耐药后的伪肿瘤样本。为了使用scRNA-seq数据确定精心整理的LIRICS相互作用的“激活”或“非激活”状态,我们如前所述采用我们的专有R工具SOCIAL来识别激活的配体-受体相互作用。符合经验标准的p值小于0.05且配体和受体的平均表达水平均超过所有样本中的中位数水平(类似于LIRICS方法)的情况被分类为“激活”。
IRIS概要
IRIS(免疫治疗耐药细胞-细胞相互作用扫描仪)作为一种监督机器学习算法运行,遵循两步过程:1. 在治疗前和治疗后非应答者组之间进行差异激活分析,以提取一组特定于细胞类型的配体-受体相互作用;2. 使用 hill climbing 聚集特征选择算法进一步优化第一步中选定的相互作用,在治疗前阶段最大化其区分应答者与非应答者的分类能力。这些步骤共同旨在推断出一组“耐药下调相互作用”(RDI)或“耐药上调相互作用”(RUI)。当有多个训练队列可用时,用于第一步和第二步的训练队列是互斥且迭代交换的,最终产生针对给定测试队列的一系列推断出的 RDI 或 RUI 的集成模型。
第一步中的差分激活分析
IRIS的第一步旨在确定在免疫检查点阻断(ICB)耐药性出现后“上调”或“下调”的相互作用。这涉及到使用特定细胞类型的配体-受体相互作用作为特征,并且目标是识别治疗前和无应答者(RECIST标准:病情稳定/进展)样本之间差异激活的相互作用。此步骤既建立了ICB耐药性后的相互作用方向,也减少了后续第二步的搜索范围。
训练涉及利用LIRICS直接输出的细胞类型特异性配体-受体相互作用活性谱。当有多个训练队列可用时,会合并这些活性谱,包括每个队列内推断出的相互作用的并集。初始特征空间可以包含多达3776种潜在的配体-受体相互作用,在黑色素瘤TME中的十种细胞类型之间产生,数据来源于LIRICS整理过的数据库。使用费希尔精确检验来识别在治疗后无应答组或治疗前组中显著激活(每个细胞对FDR < 0.2)的相互作用,并将所选择的相互作用分别归类为“上调”或“下调”的耐药性。步骤1的输出是一个根据FDR值排序的列表,其中包括被分类为RUIs或RDIs的相互作用。这些相互作用构成了步骤2中的搜索空间。
hill climbing聚合特征选择以最大化患者对ICB反应分类的交互作用在步骤2中
IRIS的第二步旨在识别一组在决定ICB反应中具有功能相关性的最优相互作用。这包括从上一步中确定的相互作用列表(RDI或RUI)作为特征开始,并且目标是选择一组能够最大化区分治疗前应答者(RECIST标准:部分/完全反应)和非应答者(RECIST标准:稳定/进展性疾病)分类能力的最佳相互作用。第二步的方法是由王等人之前工作中概述的特征选择方法组件合并而成。15和Auslander等人。9.
该算法包含一个迭代过程,由三个连续阶段组成。首先,训练队列中的预处理样本被随机分成三部分:两部分用于训练,一部分用于测试。其次,在一个空集的基础上,通过贪婪策略逐步添加最具区分性的相互作用(根据步骤1中每个细胞对的FDR排名),直到在训练折上的ICB反应分类精度(以AUC衡量)不再提高为止。考虑到阶段1中建立的个体特征的方向性性质,我们预计对于步骤1中的RUIs,较低的激活对应于响应;相反,对于RDIs,较高的激活水平与响应相关联。第三,从训练折中选择的一组相互作用一起通过分类准确性来评估,并在测试折上打分。测试AUC≥0.6时,所有选定交互集的奖励得分为1,而测试AUC<0.4时,则对所有选定交互集进行扣分-1。介于两者之间的AUC值和所有未选特征得分均为0。这一过程重复500次迭代。
经过500次迭代后,我们对每个个体交互在所有迭代中的得分进行求和,称为特征得分。我们在整个500个解决方案中识别出特征得分最高的特征,这近似于一个最小化过拟合风险的解。在特征选择过程中,可以估计经验分布p-每个特征的值与随机机会获得的特征得分相比具有更高的特征得分所关联的价值。通过将从hill climbing特征选择方法得到的每个解决方案的值进行洗牌,并对每次迭代重复此过程1000次来完成这种估计。通过对500个解决方案执行1000次随机排列,构建了一个表示随机机会下特征得分的零分布。我们选择了具有经验上显著性的特征p值小于0.05,根据输入特征的方向性,将其视为我们的参考日摄入量(RDIs)或风险利用率(RUIs)之一。经验上的p每个特征j的值估计如下:
$${p}_{j}=\frac{1}{1000}\sum_{i=1}^{1000}1_{{{\text {null feature score}}_{ij}> {\text {observed feature score}}_{ij}}}$$
(1)
在这项研究中,在步骤2特征选择过程中排除了Auslander等人的预处理队列,原因是响应者数量有限,这阻碍了可靠预测准确性的确定。
计算RDS和RUS
在黑色素瘤中识别到药物反应下调因子(RDI)或药物反应上调因子(RUI)后,给定肿瘤样本的抵抗性下调评分(RDS)和抵抗性上调评分(RUS)的计算如下进行:
$${\mbox{RDS或RUS}}=\frac{{\sum }_{i=1}^{n}\frac{{{{\rm{number}}}} \, {{{\rm{of}}}}\, {{{\rm{active}}}}\, {{{\rm{inferred}}}}\, {{{\rm{RDI}}}} \, {{{\rm{or}}}}\, {{{\rm{RAI}}}}_{i}}{{{{\rm{total}}}}\, {{{\rm{number}}}}\, {{{\rm{of}}}}\, {{{\rm{inferred}}}}\, {{{\rm{RDI}}}}\, {{{\rm{or}}}}\, {{RAI}}_{{{\rm{i}}}}}}{n}$$
(2)
首先计算每个集合的RDIs或RUIs(i)在整体模型中推断出的激活交互作用的比例。然后计算所有(n)个集合中的RDIs或RUIs在整个模型中激活交互作用比例的平均值。最终的RDS和RUS评分被调整以进行跨队列比较。
通过结合所有最初为每个队列推断出的个体RDI集成模型(Gide等,Liu等,Auslander等,Riaz等和PUCH),也导出了一个合并的集成模型。这个统一的合并模型应用于两个独立的患者队列:TCGA-SKCM和Jerby-Arnon等。
计算优势比
为了为每个测试队列建立区分真实应答者和非应答者与假阳性和假阴性的恰当分类阈值,我们考虑了推断出的RDIs集以及从步骤2中用于训练的每个性别治疗前样本集中提取的肿瘤样本。对于每个训练队列,计算每个样本中激活的推断相互作用的比例,并将其扩展到所有肿瘤样本上。利用R语言中的cutpointr包(版本1.1.2)中的cutpointr()函数以及训练集内每个肿瘤样本的反应信息,我们使用扩展后的分数计算出最佳最大化切点。56最终阈值通过计算每个训练队列单独计算的阈值的平均值来获得。
与其它最先进的ICB预测器进行基准测试
为了评估IRIS的性能,我们按照原始方法计算了每个最先进的免疫细胞浸润预测器在每个肿瘤样本中的相应得分。T细胞耗竭(Tex)特征通过平均特征中可用基因的表达来计算,包括LAG3, TIGIT, PDCD1, PD1, HAVCR2, TIM3,和 CTLA4TIDE评分是使用在线工具计算的(http://tide.dfci.harvard.edu). Ress 签名是使用作者提供的原始发布代码计算得出的,该代码可从 中获取。https://github.com/livnatje/ImmuneResistance melanocyte塑性评分(MPS)10IFNG信号印记57细胞毒性特征signature11T细胞炎症型GEP(Tin)58以及IMPRES9我们根据原始出版物中详细描述的方法计算了签名得分。
空间RNA测序数据集
我们从Thrane等人的研究中收集了两个公开可用的转移性黑色素瘤空间转录组学数据集。37以及Biermann等人。36分别来自legacy和SlideSeqV2平台。Thrane等人的研究包括未经治疗的黑色素瘤淋巴结转移的切片(n=8)。Biermann等包含了未经治疗的黑色素瘤脑转移的幻灯片(n等于9),未经治疗的颅外黑色素瘤转移病灶(n等于2),以及抗CTLA4治疗后的颅外黑色素瘤转移灶(n= 2)。两个治疗后的样本来源于对ICB方案无响应(RECIST标准:病情进展)的患者。由于空间坐标覆盖不足或之前接受过适应性T细胞疗法,我们排除了Biermann等人研究中的切片MBM07.1、MBM13.1和ECM08.1。此外,我们收集了Biermann等人所有单核RNA测序(snRNA-seq)数据,这些数据来自被分析的转移性黑色素瘤样本。n=28). 从Thrane等人的遗留数据中下载了数据(https://www.spatialresearch.org/资源和已发布数据集/),而Biermann等的SlideSeqV2和snRNA-seq数据从GEO(GSE185386)下载。
利用CytoSPACE将单细胞转录组与空间转录组对齐
使用SPECIAL(见下文)来推断空间数据中配体-受体相互作用的活性时,我们首先利用CytoSPACE(v1.0.6)将空间转录组学(ST)数据解卷积到单细胞分辨率。38 (https://github.com/digitalcytometry/cytospaceCytoSPACE 需要两个输入:1. 空间转录组学载片的计数矩阵,2. 带有细胞类型注释的参考单细胞转录组数据集的计数矩阵。它输出去卷积后的空间转录组数据,并为来自参考 scRNA-seq 数据集的选择单个细胞分配空间坐标。每个坐标处每种细胞类型的细胞丰度被计算为该特定区域内的细胞比例。
对于Thrane等人提供的大量ST数据(使用legacy平台),我们将空间点与未接受治疗样本的单细胞RNA测序数据(以计数形式)进行了分配。n来自Jerby-Arnon等人的数据(n = 16)12作为参考。细胞类型限于我们单细胞分析之前提到的十种指定的细胞类型。为了优化CytoSPACE在Jerby-Arnon等人的smart-seq计数数据上的性能,我们使用Spatial Seuart估计了每张切片的细胞比例组成。59作为用户提供的细胞分数输入用于CytoSPACE。此外,我们根据Vahid等人的指导将几何形状设置为方形,关闭降采样,并将平均细胞数设置为20,当将CytoSPACE应用于传统空间平台和smart-seq参考数据时。38.
对于Biermann等人提出的SlideSeqV2平台(即单细胞分辨率的空间转录组学),我们首先使用RCTD为每个空间位点分配单独的细胞类型。60来自spacexr包中的双胞胎模式下的pipelineR每个SlideSeqV2切片均使用所有转移性黑色素瘤snRNA-seq数据进行标注n等于28)并带有细胞类型注释。来自snRNA-seq数据的细胞类型手动重新注释如补充表所示。1双倍体细胞、“低质量”细胞和循环细胞被排除在外。RCTD的输出包括所有空间点的第一个细胞类型注释,以及仅在RCTD将空间点推断为“确定的双倍体”时才提供第二个细胞类型注释。RCTD框架借鉴了Biermann等人的方法。36接下来,我们针对Biermann等人提供的每个SlideSeqV2幻灯片执行了两次CytoSPACE(单细胞模式),仅使用匹配患者的snRNA-seq数据作为参考。第一次运行包含了所有空间点及其从RCTD获得的第一种细胞类型注释作为用户输入,而第二次运行则局限于“双倍确定”的空间点及其相应的第二种细胞类型注释。在运行CytoSPACE之前,排除了既不在匹配患者的空间转录组学幻灯片中也不在snRNA-seq中的细胞类型。
利用SPECIAL从单细胞转录组学对齐到空间转录组学中推断激活的细胞类型特异性配体-受体相互作用
为了量化每个空间转录组学载片上特定细胞类型特异性配体-受体相互作用的活性,我们将内部开发的单细胞配体-受体推断工具SOCIAL进一步发展为SPECIAL(SP单细胞转录组学 cEll-Cell 我交互AL算法;见补充图(fig.)12此版本专门针对空间转录组学进行了定制,适用于与单细胞转录组数据对齐的情况,CytoSPACE的直接输出结果(见上文)。SPECIAL的主要三个步骤概述如下(参见补充图)。12).
在第一步中,将每个载玻片的空间坐标划分为直径约为250μm的空间“区域”,这一距离有利于旁分泌配体-受体相互作用(基于文献)。39,通过两种可能的方法。对于批量空间转录组平台(即传统平台或Visium 10X),我们开发了一种滑动窗口方法。这种方法在每个标记的空间坐标内将一定半径范围内的细胞进行合并。区域的数量取决于给定载片上测序的空间坐标的数量,每个区域的空间覆盖可以与其他多个区域重叠。对于SlideSeqV2平台(或其他具有圆形测序区域或“puck”的单细胞分辨率空间转录组平台),我们开发了一种基于K-means聚类的方法。该方法根据指定直径将单个细胞的x和y坐标聚类为不重叠的圆形区域。这里,区域的数量由指定直径的圆区域数量决定。i直径为的圆形序列区域中可以容纳的j对于这两种方法,仅含有一种细胞类型的区域都不会被纳入后续分析。
在这项研究中,我们对Thrane等人(Legacy平台)的研究队列采用了滑动窗口方法,将每个1单位半径内的细胞(最大直径为300微米的区域)合并在一起。而对于Biermann等人的研究队列(SlideSeqV2平台),我们使用了K-means聚类方法,将每张切片划分为144个区域。选择144个区域是基于在SlideSeqV2平台上用于测序的直径为3000微米的圆形“puck”内可以容纳多少个直径为250微米的圆形区域而定。
在第二步中,为了辨别每个空间区域内精选的LIRICS相互作用是“激活”还是“非激活”的状态,我们使用了之前描述的专有R工具SOCIAL。具体来说,SOCIAL独立应用于每个区域,包括相应的单细胞转录组。
在第三步中,与实证数据的交互p值小于0.05且配体和受体的平均表达量均高于所有区域中的中位数(类似于LIRICS)的过程被归类为“激活”。
报告摘要
有关研究设计的进一步信息可在以下位置获得:自然科研报道摘要与此文章相关的内容。
数据可用性
Gide等人的bulk RNA-seq数据、CODEFACS的去卷积表达和细胞分数数据16里亚兹等人。18刘等人。17王等人中的TCGA-SKCM15可通过Zenodo仓库获取61 (https://zenodo.org/records/5790343TCGA-SKCM的生存时间线可从UCSC Xena浏览器获取。53 (https://xenabrowser.net病理分类可参考Saltz等人的研究。33Auslander等人的bulk RNA-seq数据9可以从GEO通过登录号访问GSE115821以及对于PUCH19从GitHub(https://github.com/xmuyulab/ims_gene_signature杰伯里-阿农等人的单细胞RNA测序数据12可以从GEO通过登录号访问GSE115978,带有从TISCH2获取的额外单元元信息62 (http://tisch.comp-genomics.orgThrane等人的空间RNA测序数据37可从(中获取https://www.spatialresearch.org/资源-发布的数据集/对于Biermann等人。36,单核和空间RNA测序数据均可从GEO(基因表达数据库)中通过访问号获取。GSE185386本研究生成的CODEFACS、LIRICS、SOCIAL和SPECIAL数据(及相关输入)已存放在Zenodo存储库中。63 (https://zenodo.org/records/13172848). 本论文提供了源数据。其余数据可在文章、补充信息或源数据文件中获取。源数据本文提供了这些内容。
代码可用性
本研究的结果再现所需的工具(IRIS、SOCIAL和SPECIAL)和代码可通过GitHub获取。https://github.com/KWangLab/NatCommun_Sahni2024去卷积工具CODEFACS和LIRICS可通过Zenodo仓库获取。https://zenodo.org/record/5790343CytoSPACE可通过GitHub获取https://github.com/digitalcytometry/cytospace).
参考文献
白瑞,吕泽,徐迪及崔 الج (Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. 中文翻译未能找到具体人名对应的中文名字,因此保留原文不变)免疫检查点抑制剂癌症免疫治疗的预测生物标志物。生物标记. 研究. 生物医学中心 8, 34 (2020).
莫拉德 G.,赫尔明克 B. A.,沙玛 P.,瓦戈 J. A. 免疫检查点抑制的应答、耐药和毒性的特征。细胞杂志。184, 5309–5337 (2021).
黄安昌 & 拉斐尔·扎帕索迪,黑色素瘤免疫检查点阻断免疫治疗十年:理解免疫敏感性和耐药性的分子基础。自然·免疫学 23, 660–670 (2022).
夏尔马,P.,胡-利斯科万,S.,沃戈,J. A. & 里巴斯,A. 原发性、适应性和获得性癌症免疫疗法耐药性。细胞 168, 707–723 (2017).
费斯,C. M., 范艾伦,E. M., 德雷克,C. G., 阿林森,J. P. & 哈利斯科万,S. 免疫检查点阻断耐药机制:为什么免疫检查点抑制剂免疫疗法对所有患者均无效?美国临床肿瘤学会教育手册 39, 147–164 (2019).
安德森,N. M. & 西蒙,M. C. 肿瘤微环境。当代生物学30R921–R925 (2020)
史密斯,M. J.,恩 Gow,S. F.,里巴斯,A. &滕,M. W. L. 针对肿瘤微环境的组合癌症免疫疗法。自然综述·临床肿瘤学 13, 143–158 (2016).
江,P.等. T细胞功能障碍和排除的标志预测癌症免疫治疗反应。自然医学. 24, 1550–1558 (2018).
Auslander, N. 等人. 对转移性黑色素瘤免疫检查点阻断疗法反应的稳健预测。自然医学 24, 1545–1549 (2018).
佩雷斯-吉哈罗,E. 等。多模型临床前平台预测免疫治疗对黑色素瘤的临床反应。自然医学杂志. 26, 781–791 (2020).
Davoli, T., Uno, H., Wooten, E. C. & Elledge, S. J. 肿瘤非整倍体与免疫逃避标志物相关,并且对免疫治疗的反应减弱。 Sci. 355,eaaf8399(2017)。
Jerby-Arnon, L.等。一种癌细胞程序促进T细胞排除并导致对检查点抑制的抵抗。细胞 175, 984–997 (2018).
张亚平等。与多种肿瘤类型免疫治疗反应相关的T细胞韧性模型。自然医学杂志 28, 1421–1431 (2022).
阿林戈尔, E., 艾弗里, A., 哈里斯门迪, O. & 刘易斯, N. E. 从基因表达解析细胞间相互作用和通讯。自然遗传学评论 22, 71–88 (2021).
王凯等。利用CODEFACS和LIRICS从bulk基因表达中解卷积临床相关的细胞免疫互作,将黑色素瘤患者分类为抗PD-1疗法响应者。癌症发现杂志 12, 1088–1105 (2022).
盖德,T. N. 等人。不同的免疫细胞群定义了抗PD-1单药治疗和抗PD-1/抗CTLA-4联合治疗的反应。癌细胞 35,238-255.e6 (2019).
刘,D.等.将临床结局与PD1阻断整合的分子和临床模型在转移性黑色素瘤患者中的应用。自然医学 25, 1916–1927 (2019).
里亚兹,N.等. 使用尼伏单抗免疫治疗期间肿瘤和微环境的演变。细胞 171, 934–949.e16 (2017).
崔等人。比率干扰素-γ信号与免疫抑制信号的比值可预测黑色素瘤抗PD-1治疗反应。NPJ基因组医学 6, 7 (2021).
Kumar, M. P. 等人。单细胞RNA测序分析识别出与肿瘤特征相关联的细胞间通信。细胞报。注意:"Cell Rep."通常指的是学术期刊《Cell Reports》,在正式文档中应保持其英文原名或根据具体语境使用"《细胞报告》"这样的标准译法。此处直接翻译可能不完全准确,建议保留原文形式或采用标准译名。如果必须翻译,则给出的“细胞报”是一个直译结果。 25, 1458–1468(2018).
Vento-Tormo, R. 等人. 人类早期母胎界面的单细胞重建。自然的 563,347–353 (2018). 可从以下链接获取。
哈林,H.等.与CD8 + T细胞招募相关的黑色素瘤转移灶中的趋化因子表达。癌症研究. 69, 3077–3085 (2009).
梅西纳等人。在黑色素瘤中鉴定出类似淋巴结的结构的化学因子基因特征(12-chemokine基因签名):对免疫治疗患者选择的潜在作用?科学报告 2, 765 (2012).
奥兹加,A. J., 周,M. T. & 赖斯泰尔,A. D. 化学趋化因子与癌症免疫反应。免疫学 54, 859–874 (2021).
Reschke, R. 等人。免疫细胞和肿瘤细胞产生的CXCL10是转移性黑色素瘤对免疫治疗反应的指示标志。免疫治疗癌症期刊 9,e003521 (2021).
李旭,戴鹤,王华,韩伟。探索癌症免疫治疗中的先天免疫:机遇与挑战。细胞分子免疫学 18, 1607–1609 (2021).
吉内弗拉·P., 洛鲁索·G., 瓦尼尼·N. 固有免疫细胞及其对基于T细胞的免疫治疗的贡献。国际分子科学杂志. 21, 4441 (2020).
科赫利,K.,皮拉里塞蒂,V. G. & 金,T. S. 关键趋化因子引导实性肿瘤中免疫细胞的迁移。癌症基因治疗杂志 29,10–21(2022)。Available from.
邹,M. T. 等。肿瘤内的CXCR3趋化因子系统的活性是抗PD-1疗法有效的必要条件。免疫学 50, 1498–1512 (2019).
巴里,K. C. 等人。天然杀伤细胞-树突状细胞轴定义了对检查点疗法有反应的肿瘤微环境。自然医学 24, 1178–1191 (2018).
张艳,关雪艳及江鹏.肿瘤中T细胞排除的细胞因子和趋化因子信号。免疫学前沿杂志 11, 594609 (2020).
里士满,A.,杨,J.及苏,Y.趋化因子/趋化因子受体在黑色素瘤中的利与弊。色素细胞黑色素瘤研掙(注:「Res」可能是「Research」的缩写,但此处中文通常不保留该英文词) 如果必须只输出实际可翻译部分且保持简洁,则为: 色素细胞黑色素瘤研究 22, 175–186 (2009).
Saltz, J. 等人。利用病理图像深度学习技术对肿瘤浸润淋巴细胞的空间组织和分子相关性进行研究。细胞报告(Cell Reports) 23, 181–193 (2018).
Thomas NE 等人. 原发性黑色素瘤中肿瘤浸润淋巴细胞分级在基于人群的基因、环境和黑色素瘤研究中与黑色素瘤特异性生存独立相关。临床肿瘤学杂志 31, 4252–4259 (2013).
克莱门特 C. G. 等人。原发性皮肤黑色素瘤垂直生长期肿瘤浸润淋巴细胞的预后价值。癌症. 77, 1303–1310 (1996).
Biermann, J. 等人. 分解人类黑色素瘤脑转移的治疗初治生态系统。细胞 185, 2591–2608 (2022).
Thrane, K., Eriksson, H., Maaskola, J., Hansson, J. & Lundeberg, J. 空间分辨转录组学能够解析III期皮肤恶性黑色素瘤的遗传异质性。癌症研究78, 5970–5979 (2018).
瓦希德,M. R. 等人。利用CytoSPACE对单细胞和空间转录组进行高分辨率对齐。自然·生物技术 41, 1543–1548 (2023).
弗朗西斯,K. & 帕尔松,B. O. 细胞间有效通信距离由细胞因子和化学趋化因子分泌与扩散的相对时间常数决定。 Proc. Natl Acad. Sci. 94, 12258–12262 (1997).
弗里德曼 W. H., 派热斯 F., 萨特丝-弗里德曼 C. & 加隆 J. 人类肿瘤中的免疫微环境:对临床结局的影响。自然·癌症综述 12, 298–306 (2012).
刘,Z等.CXCL11武装的溶瘤痘病毒引发强烈的抗肿瘤免疫并表现出增强的治疗效果。肿瘤免疫学 5吴1091554(2016).
孙昱,Finger查尔斯,阿尔瓦雷斯-瓦利尼亚莱昂,齐楚特克克laus,布赫霍尔茨克里斯蒂安。通过复制型逆转录病毒载体慢性递送干扰素诱导蛋白10抑制肿瘤生长。癌症基因治疗杂志 12, 900–912 (2005).
Feldman AL 等。逆转录病毒基因转移干扰素诱导蛋白10抑制人黑色素瘤异种移植的生长。国际癌症杂志 99, 149–153 (2002).
Arenberg, D. A., White, E. S., Burdick, M. D., Strom, S. R. & Strieter, R. M. 在用干扰素-γ诱导蛋白10(IP-10/CXCL10)治疗的肿瘤携带SCID小鼠中生存率提高。癌症免疫学,免疫治疗学 50, 533–538 (2001).
王旭,吕晓莉,赵慧艳,张凤春及江小斌。IP10-EGFRvIIIscFv重组蛋白与CD8+细胞毒性T淋巴细胞协同抑制植入小鼠胶质瘤生长。癌症免疫学,免疫治疗学 62, 1261–1272 (2013).
潘俊等。CXCR3/CXCR3配体生物学轴通过免疫血管稳态机制抑制RENCA肿瘤生长。免疫学杂志 176, 1456–1464 (2006).
张瑞等。MIG(CXCL9)趋化因子基因疗法与低剂量顺铂联合使用可提高对小鼠癌瘤的治疗效果。基因治疗 13, 1263–1271 (2006).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet:通过将配体与靶基因相连来建模细胞间通讯。自然方法杂志 17, 159–162 (2020).
Choi H.等. 单个间质细胞群体的转录组分析揭示了小鼠肺癌模型中的间质-肿瘤相互作用。细胞报告(Cell Reports) 10, 1187–1201 (2015).
Sheinin, R. 等人。interFLOW:用于识别介导多种受体信号汇聚的因素的最大流框架。npj系统生物学与应用 10, 66 (2024).
Russo, E., Santoni, A. & Bernardini, G. 肿瘤抑制还是肿瘤促进?CXCR3在癌症中的双重作用。J. 白细胞生物学 108, 673–685 (2020).
张,T.等.CX化学趋化因子受体4和CX化学趋化因子配体12在T细胞向黑色素瘤细胞迁移中的优先作用。癌症生物学与治疗学 5, 1304–1312 (2006).
Goldman, M. J. 等人通过Xena平台可视化和解释癌症基因组数据。自然·生物技术 38, 675–678 (2020).
梅拉德,M.,萨特,P.,赫尔夫特,J.,米勒,J.及莫尔塔,A. 树突细胞谱系:稳态和炎症状态下树突细胞及其亚群的发育和功能。免疫学年报 31, 563–604, (2013).
科林, M. & 贝格利, V. 人类树突细胞亚群:最新进展。免疫学 154, 3–20 (2018).
Thiele, C. & Hirschfeld, G. cutpointr:在R中改进的最佳切点的估计和验证。统计软件期刊 98, 1–27, (2021).
Ayers, M. 等人。IFN-γ相关的mRNA谱型可预测PD-1阻断的临床反应。J.临床研究 127, 2930–2940 (2017).
斯坦因切,T. 等人。食管癌患者T细胞炎症基因表达谱和PD-L1表达的预后意义。癌症医学 10, 8365–8376 (2021).
斯图尔特等人。单细胞数据的全面整合。细胞 177, 1888–1902, (2019).
Cable, D. M. 等人。空间转录组学中细胞类型混合物的稳健分解。自然·生物技术 40, 517–526 (2022).
王K.等。利用CODEFACS和LIRICS从总体基因表达中解析临床相关的细胞免疫互作,对黑色素瘤患者进行抗PD-1治疗分层。预印本于Zenodo; https://doi.org/10.1101/2021.01.20.427515 (2021).
韩 Y. 等人. TISCH2:扩大数据集和新的工具用于肿瘤微环境单细胞转录组分析。核酸研究[Nucleic Acids Res互联网版] 51D1425–D1431 (2023).
萨尼 S. 等。一项机器学习模型揭示了在对免疫检查点阻断产生耐药性的黑色素瘤中,配体-受体相互作用的广泛下调增强了淋巴细胞浸润。预印本发表于Zenodo; https://doi.org/10.1101/2023.09.20.558683 (2024).
致谢
本研究部分得到了美国国立卫生研究院内部研究计划、国家癌症研究所的支持。该工作利用了美国国立卫生研究院HPC Biowulf集群的计算资源http://hpc.nih.gov此处的结果部分基于TCGA研究网络生成的数据:https://www.cancer.gov/tcga此外,我们还想感谢癌症数据科学实验室成员对此工作的有益反馈。图.figure 1图B面板,Fig.3图D面板,Fig.6补充图 fig.11以及补充图 fig.12由BioRender.com创建,根据创用CC 姓名标示-非商业性-禁止改作 4.0 国际许可协议发布。
资金
美国国立卫生研究院提供了开放访问基金。
伦理声明
利益冲突
E.R.是Medaware有限公司、Metabomed有限公司和Pangea生物医药有限公司(后者已剥离)的联合创始人。E.R.担任Pangea生物医药有限公司无薪科学顾问。其余作者声明他们不存在潜在的利益冲突。
同行评审
同行评审信息
自然通讯感谢秦马及其他匿名审稿人对本工作的评审。审稿文件可用。
附加信息
出版者 notes施普林格·自然对于出版地图和机构 affiliation 中的管辖权主张保持中立。注意这里的affiliation通常译为“隶属关系”或“从属关系”,但此处直译保留原意。
补充信息
权利与许可
开放存取本文采用知识共享署名4.0国际许可协议进行许可,该协议允许任何媒体或格式下的使用、分享、改编、分发和复制,只要您适当引用原始作者和来源,并提供指向知识共享许可的链接,同时指明是否进行了修改。除非材料的版权信息中另有说明,本文中的图片或其他第三方材料均包含在文章的知识共享许可范围内。如果材料未包含在文章的知识共享许可内且您的使用意图不受法律规定允许或超出允许范围,则您需要直接从版权所有者处获得许可。要查看该许可证副本,请访问http://creativecommons.org/licenses/by/4.0/.
关于这篇文章

引用这篇文章
沙尼,S.,王,B.,吴,D.等式中的其他人或作品中的其他作者等人(通常用于引用多作者文献)一个机器学习模型揭示了在对免疫检查点阻断产生耐药性的黑色素瘤中,配体-受体相互作用的广泛下调,这些下调增强了淋巴细胞的浸润。自然通讯 15,8867 (2024). https://doi.org/10.1038/s41467-024-52555-4
收到:
已接受:
发布:
DOI: https://doi.org/10.1038/s41467-024-52555-4