

人工智能蛋白质结构预测和设计获得诺贝尔奖
作者:Jamie Durrani
大概三十分钟前,我转向我的妻子说:“我想今年是不会有了。”……然后三十秒后,我接到了来自瑞典的电话。
等待今年诺贝尔化学奖的宣布时,约翰·贾姆珀认为自己获奖的概率是“十分之一”。许多其他观察者则会认为他的获奖几率要高得多,因为贾姆珀及其在谷歌DeepMind的同事所创建的AlphaFold平台产生了巨大影响。AlphaFold准确预测蛋白质三维结构的能力已被多次描述为生物化学领域的革命,诺贝尔委员会在宣布贾姆珀和DeepMind首席执行官德米斯·哈萨比斯获得今年奖项的一半时也表达了同样的观点。
奖金的另一半授予了蛋白质设计先驱大卫·贝克,他的美国华盛顿大学实验室开启了一种全新的非天然蛋白质领域。
DeepMind团队和Baker实验室开发的工具利用人工智能解决了两个长期以来吸引科学家的问题:如何仅根据氨基酸序列确定蛋白质的结构,以及如何创造具有自然界前所未见形状和功能的新蛋白质。
一个久经多年的难题
每一个蛋白质都是由一条氨基酸链折叠成的独特扭曲和缠绕结构组成的。这种形状对于蛋白质的生物活性至关重要,决定了它将如何与其他接触的生物分子相互作用。常见的特征包括称为α-螺旋的螺旋状部分以及一些平行走行形成较大单元(称为β-折叠)的部分。
在现实生活中,蛋白质只需几秒钟就能折叠成它们优选的构象。
在20世纪中叶,美国马里兰州的国立卫生研究院的主要化学生物学家克里斯蒂安·安芬森使用一种名为牛胰核糖核酸酶的蛋白质进行了一系列开创性的实验。他证明了如果让核糖核酸酶从其自然结构展开并随后允许它重新折叠,它总会恢复到原来的形状。这使安芬森得出结论,蛋白质的天然状态是由氨基酸的序列决定的这一发现使他获得了1972年诺贝尔化学奖的一半奖项。
不久之后,美国麻省理工学院的分子生物学家Cyrus Levinthal观察到,即使是一个相对短小的蛋白质也可能潜在地采取一个无法想象的巨大数量的不同构象。如果未折叠的蛋白质每秒随机采样数万亿种可能的状态,它仍然需要极长的时间才能尝试所有这些状态。但在现实生活中,蛋白质只需几秒钟就能折叠成其首选的构象。
这导致莱文塔尔认为结论蛋白质折叠是由氨基酸链上的局部相互作用“加速和引导”的,这些相互作用启动了折叠过程。
安芬森和莱文thal的发现激发了生物学的一项重大挑战:如果指导蛋白质折叠所需的所有信息都存储在氨基酸序列中,那么一定有一种方法可以破解这一密码。这样科学家就能够预测蛋白质所偏好的形状,为研究蛋白质的功能提供新的途径,并加速科学突破。研究人员将摆脱通过晶体实验诱导蛋白质形成晶体并对其进行衍射分析的繁琐工作。
结构预测的世界杯
“很多对计算感兴趣的人抓住了这个想法,”他说。约翰·莫尔特美国马里兰大学的一名计算生物学家说:“从上世纪70年代起,整个行业就开始为此而形成了。”
我震惊地发现要判断某件事是否有效是多么难。
但莫尔特解释说,当这些努力开始时,并没有很好的方法来评估不同的方法。‘当我进入计算领域时,我震惊地发现很难判断某件事情是否有效。很明显,我们并没有严格审视我们的工作方式,也没有充分评估:好吧,我尝试了这个假设——它是否有效?’
1994年,Moult和他的同事Krzysztof Fidelis发起了一个新的名为“蛋白质结构预测关键评估”(Casp)的项目。这个概念本质上是一个每两年举行一次的蛋白质结构预测“世界杯”:最好的团队使用他们的方法来预测选定的一组蛋白质的结构,并通过将其结果与未公布的晶体学数据进行比较来进行评分。
贝克实验室在1998年首次使用了Casp的第三个版本。他的团队的方法是基于分析长度小于10个氨基酸残基的短蛋白质序列,并利用这一点来模拟蛋白质中可能出现的常见局部相互作用。他们还考虑到了较远距离的相互作用,如主链氢键以及疏水侧链倾向于定位在蛋白质核心的趋势。该程序被称为罗塞塔计划在破解埃及象形文字的石碑之后。
罗塞塔程序将不断开发和改进,成为未来Casp竞赛中表现最佳的软件包之一。
按设计的蛋白质
在Casp3上首次亮相后,贝克实验室继续进行蛋白质结构预测的工作,但也开始了一项新的研究,将问题完全反过来思考:如果你希望某个蛋白具有特定的形状,那么需要什么样的氨基酸序列?

当我刚开始这里的工作时,我非常专注于尝试理解蛋白质折叠的原则,然后利用这些原则开发用于描述折叠过程的模型,并预测蛋白质结构,贝克回忆道。“所以我们真正开始进行蛋白质设计的工作大约是在20世纪90年代末,当Brian Kuhlman来到我的实验室的时候。”
库尔曼,现任美国北卡罗来纳大学教堂山分校蛋白质设计实验室负责人的人于1999年作为博士后加入贝克的团队。两年前,加州理工学院的研究人员报告了合成成果首个完全由计算机设计的蛋白质– 一个被称为FSD-1的由28个氨基酸组成的序列。该蛋白质复制了一个天然存在的结合锌的蛋白质的形状,而无需任何金属离子来稳定结构。
受那项工作的启发,Kuhlman 热衷于专注于设计问题。“当我去西雅图大卫实验室面试时,我记得告诉过他我对蛋白质设计的兴趣……而他也非常兴奋能进入这个领域,” Kuhlman 说。“我还记得从一开始我们就觉得‘哇,我们应该尝试设计一些前所未见的东西——那会真的很酷。’”
Kuhlman 开始用 Fortran 编程开发方法,几个月后团队就拥有了一个工作程序,这个程序可以根据蛋白质的主链预测出一种有利的氨基酸序列。“接下来面临的重大挑战是如何为全新的折叠结构创建这种新颖的主链,”Kuhlman 说。“于是我们开始将 Rosetta 的结构预测能力与这一序列设计程序结合起来。”
“aha”时刻
研究团队确定了一个由93个氨基酸残基组成的靶点,并将其命名为Top7。Top7蛋白的独特之处在于,它被故意设计为避免采用记录在蛋白质数据银行中的折叠模式和氨基酸序列——这是一个实验性蛋白质结构的存储库。换句话说,Top7是一个全新的蛋白质与自然界此前发现的任何蛋白质都无关。
贝克实验室的另一位研究人员,古塔姆·达塔斯,现在在美国圣路易斯华盛顿大学领导一个小组的这位研究人员当时负责在细菌中表达该蛋白质并将其分离出来。Top7随后通过X射线晶体学进行了表征,得到了来自巴里·斯托达德在美国西雅图的弗雷德哈钦森癌症中心。
当我们在解析晶体结构时,它几乎与设计模型完全一致,贝克回忆道。“那是非常令人兴奋的时刻——当我们看到晶体结构如此完美地匹配时,真是相当令人惊讶。”
当晶体学结果出来时,Kuhlman 已经离开 Baker 的实验室,在北卡罗来纳大学开始了自己的研究小组。“我收到了一个包含蛋白质坐标信息的电子邮件,我可以将其在查看器中打开并与设计模型进行叠加。两者之间有着美妙的一致性,”他说。“我认为这可能是我的科学职业生涯中最开心的一天,因为那一刻就像是‘啊哈!’的顿悟时刻。”
巴克尔证明了设计全新蛋白质是可能的之后,他开始考虑下一个问题。他指出:“Top7没有任何功能——它是完全惰性的。”因此,接下来的挑战就是:我们如何设计能够实际发挥作用的蛋白质?
2008年,该小组生产了显示酶活性的一组蛋白质这些蛋白质被精心设计,以具备能够促进底物分子发生逆式 aldol 反应的活性位点,而这种底物分子在自然界中是不存在的。尽管这些酶表现出的活性相对较低,但该项目标志着功能性蛋白质设计新时代的开始。
幸运转盘
那一年,约翰·詹珀放弃了物理学的博士学位,并在考虑下一步该做什么。他说:“作为一名失业的物理学家,我做了人们通常会做的事情,申请了金融行业的工作。”“在我生命中的一次幸运转折中,我的申请从对冲基金D E Shaw被转到了进行分子动力学研究的D E Shaw Research小组。”
那是我第一次接触生物学。而我很快就爱上了它,因为我在解决非常有趣且困难的计算问题,但同时我也能够看到这些问题与疾病和健康之间的深远影响。

在与shaw合作三年后,经历了他首次涉足蛋白质折叠领域 jumper 返回了研究生院。这次他搬到美国的芝加哥大学,在一位生物化学家的指导下工作。托宾·索斯尼克理论化学家卡尔·弗里德.
“他们有一个联合实验室,他们在折叠动力学和蛋白质折叠路径问题等多个方面进行了研究,”Jumper说。“我最初加入这个实验室,并不是因为它是专注于蛋白质折叠的实验室,而是因为它是一个结合了实验与计算的混合实验室。当时我觉得自己需要真正理解实验人员是如何工作的,确保我不生活在计算的泡沫中度过一生。”
当Jumper在攻读博士学位时,他的未来同事Hassabis凭借他在2010年共同创立的DeepMind公司在人工智能界引起了轰动。DeepMind结合了Hassabis的两大兴趣领域——神经科学和计算机编程,致力于实现“解决智能”的终极目标,并创造真正的通用人工智能。
该公司首先开始用复古视频游戏训练其机器学习模型,证明了AI能够学会玩《乒乓球》和《太空侵略者》等游戏,并最终达到了专家级别。2016年,其产品AlphaGo通过成为围棋高手而引起了轰动。第一个击败的计算机程序顶级的职业围棋选手。
大约在这个时候,DeepMind决定将其技术转向蛋白质折叠问题。Jumper解释说,Hassabis自大学时代就一直有一个长期目标,即“利用人工智能攻克科学难题”,他早就意识到了这个问题的存在。
完全是巧合,DeepMind 每半年会有一个黑客马拉松周,在这个星期内你可以做自己想做的任何事情——而 Rich Evans 等几位仍然在 AlphaFold 团队的人当时搜索了“生物学中的重大挑战”,蛋白质折叠问题出现了,” Jumper 补充道。DeepMind 的研究人员花了一周的时间试图训练一个强化学习代理来控制 Foldit——这是一款在 2008 年由 Baker 实验室创建并通过 Rosetta 驱动的视频游戏。注册公民科学家为了帮助解决蛋白质折叠问题。 jumper指出,这与Demis长期以来的兴趣相结合,成为了AlphaFold项目。
在他攻读博士学位的最后阶段,Jumper 听到了关于刚刚起步的 AlphaFold 项目的传闻,并申请了在 DeepMind 工作。
卡斯普的惊叹声
Jumper加入公司六个月后,DeepMind团队参加了Casp13竞赛。该比赛采用一种名为全局距离测试(GDT)的评分方法,根据预测结构与蛋白质实际结构的匹配程度提供一个百分比准确度。尽管在Casp的历史进程中模型有所改进,但多年来获胜程序的GDT得分一直徘徊在30%至40%之间。
2018年,AlphaFold1宣布参加比赛得分接近60%。该系统基本上使用了图像识别软件来绘制存储在蛋白质数据银行中的蛋白质结构,然后利用这些信息训练算法预测新蛋白质的未知结构。
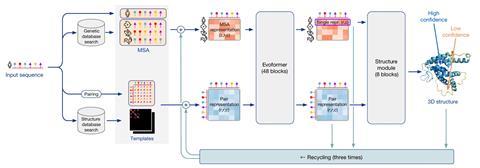
穆尔特指出,在当时,其他几个小组也采取了类似的方法,其中一些团队达到了与DeepMind团队相当的准确性。“如果你看看2018年每个目标的最佳模型是谁,DeepMind比任何其他人要好得多,但也有其他人在竞争中紧随其后——所以这虽然令人惊讶,但却不是出乎意料的事情,”他说。“2020年发生的事情则令人惊叹。”
Casp13之后不久,Jumper被任命为AlphaFold项目的负责人。他解释说,AlphaFold1采用了“接近纯粹的深度学习”的蛋白质折叠方法。“我们将现成的计算机视觉网络拿来使用,我们会对其进行良好的调整,并进行复杂的优化,”他回忆道。“我记得我当时在想,我们必须做更多的工作来引入对蛋白质物理的理解。”
团队开始测试许多不同的想法——根据Jumper的说法,“有些真的很棒,有些则很无聊”——以逐步提高AlphaFold模型的准确性。
重要的是,Jumper指出,在系统开发方式上存在一种“整体哲学上的变化”。他解释说:“对于AlphaFold1,神经网络与图像识别网络完全相同。相反,我们尝试将我们对蛋白质、进化、生物学、几何学和物理学的理解构建到神经网络的结构中。” “因此,我们开始专门为这个问题设计新的神经网络技术……而这最终产生了戏剧性的效果。”
系统生物学家后来进行的分析穆罕默德·阿尔库拉伊shi(注意:人名部分直接转写,不一定有标准的中文形式,此处为一种可能的译法)在美國哥倫比亞大學的研究會表明AlphaFold2只需要1%的训练数据从蛋白质数据银行生成更准确的预测,比原来的AlphaFold1系统。
因此,构建围绕蛋白质问题的机器学习大约相当于额外获得了100倍的数据量,这是非常明确的说法, jumper表示,“我认为整个故事是人们已经建立了图像的深度学习和文本的深度学习,但没有人去建立蛋白质的深度学习。而我认为这可能是戏剧性的差异所在。”
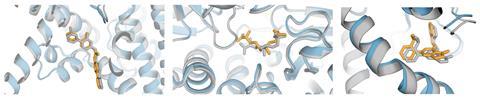
当AlphaFold2在2020年的Casp14竞赛中参赛时,它震惊了蛋白质结构预测界该系统对于难目标的平均GDT分数达到了约90%,与实验上确定蛋白质结构的方法相当。
毛尔描述了竞赛组织者在分析AlphaFold2的预测时感到的惊讶:“我们一个个地收集结果,当你看到一个难以攻克的目标时(你会想)‘哇,太棒了!’然后你看下一个:‘这简直不可思议!’再看下一个:‘到底发生了什么?人们怎么能突然用实验精度做到这一点?’”他回忆道。“所以那真是让我们震惊、兴奋和感到突破的时刻。”
AlphaFold2的结果引领了Casp14组织者声明折叠问题“对于单一蛋白质而言已被很大程度上解决”。其他研究人员也有类似的反应。AlQuraishi写道,在博客文章“我一生中从未料到会看到如此迅速的科学进步”,并补充说“核心领域已经被彻底颠覆”。
生物科学革命
自从AlphaFold2的结果公布以来,贝克实验室和DeepMind团队一直在利用该技术应对生物科学领域的主要挑战。
几乎马上,DeepMind团队就开始使用AlphaFold2来解析他们能找到的所有蛋白质的结构。在2021年,他们发布了一个包含35万个预测结构的数据库,其中包括了人体中发现的每一种蛋白质一年后,团队发布了另外2亿个结构几乎涵盖了科学界已知的所有蛋白质。
几个月内就已经有了关于AlphaFold的发现
该资源已经对全球学术界产生了立竿见影的影响,迄今为止已有超过两百万名研究人员使用了该数据库。在仅仅三年内,DeepMind团队的AlphaFold2的主要论文已被Web of Science引用了16,000次。Jumper说:“真正让我震惊的是,它已经迅速融入人们的科学研究之中——仅仅几个月后就已经有新的发现是基于它的。”
我记得这个特别刊期的科学关于核孔复合体的结构……提到了大约100次AlphaFold,而我们并没有参与——这出现在我们的论文发表一年后,”他补充说。“我认为那时候我真正意识到人们在没有我们的情况下做了多少科学研究。”
贝克的团队本身很快从DeepMind取得的进步中学习到了很多。贝克说:“当AlphaFold的结果发布时,一些资深教授感到非常沮丧和悲观,他们认为‘现在科技公司将会完全主导结构生物学领域,那么学术界还剩下什么可做的呢?’”“但我感到非常兴奋。我们只是觉得我们必须学会如何做这类事情。”
DeepMind 最终发布了AlphaFold2的源代码,但几个月来人们不清楚是否会这样。在此期间贝克的研究助理白明京现在在韩国首尔国立大学运营自己实验室的她开始着手尝试解析该程序,并将其一些底层技术应用于罗塞塔。
戴维觉得如果我们真的想将那种蛋白质结构预测方法扩展到其他应用,比如蛋白质设计上,我们就必须开发我们自己的版本——我们需要一个我们真正理解并且可以轻松根据自身需求进行修改的工具。她说。
通过结合一些关于如何将数据输入AlphaFold2以及该程序如何处理空间信息的理念,白震清迅速创建了RosettaFold实现接近DeepMind产品展示的预测准确率。重要的是,Baker团队能够利用所学的知识将其蛋白质设计工作推进到下一个层次。

团队开始构建新的AI驱动的蛋白质设计工具,这些工具显著提高了实验室旨在创建的蛋白质的复杂性和准确性。其中一个程序是生成式AI模型叫做RFdiffusion贝克解释说:“它基本上是针对蛋白质结构生成对罗塞塔折叠进行微调。”“它的效果非常好——这对于蛋白质设计来说是一个巨大的进步。”
RFdiffusion和另一个叫做的包叫蛋白质MPNN已被证明在设计能够结合其他蛋白质和分子靶点的蛋白质时特别有效。
生成式AI方法让我们能够做到的是,用一种极简的方式描述你想要的功能,然后生成式AI就会构建出具有这些特性的蛋白质结构——如果你在目标存在的情况下进行扩散过程,你会得到一个与目标完美互补形状的蛋白质,贝克说。“如果你描述了一个催化位点,那么扩散过程会围绕该位点构建一个蛋白质。因此,在此之前我们的活性很低,而现在我们直接从计算机中得到了非常高活性的结果,这真的很令人兴奋。”
这些设计蛋白质不再只是科学奇观,而是展现出解决现实世界问题的潜力。2022年,韩国批准了基于自组装蛋白质纳米颗粒的新冠疫苗在华盛顿设计的尼尔·金,巴克尔的一位前博士后研究员。与此同时,巴克尔的实验室最近发布了一篇预印本论文设计与蛇毒结合的蛋白质并似乎可以作为解蛇毒的药物。
DeepMind也在继续推出新产品。今年五月,该公司报告了其AlphaFold3模型能够预测蛋白质与其他多种生物分子(包括核酸、糖类和抗体)复合物结构的技术。该公司通过发布报告宣布了这一消息。声称AlphaFold3 “预测了所有生命分子的结构和相互作用”。
Jumper承认,由于像RNA和DNA这样的分子相对于蛋白质存在较少的训练数据,因此使用该软件完成这些任务会“更加不确定”。然而,他对公司将继续推动其软件所能实现的界限充满信心。“我们真正需要做的是改进和完善——正如我开玩笑所说的那样,更深入地学习——想出经常聪明的想法,但其实只是提高整体准确性。”他说,“然后所有这些微妙的效果,我认为最终都会水落石出。”
AI还能回答什么问题?
虽然最初的蛋白质折叠问题似乎已经有了答案,Moult 指出仍然存在新的挑战需要应对。他问道:“还有哪些其他问题可能用这些方法解决?”“最明显的一个是 RNA 结构……另一个充满炒作的领域是计算与药物相关的分子如何结合到蛋白质上。”另一个尚未完全解决的话题是能够采用多种结构的蛋白质.
许多观察者也质疑人工智能解决蛋白质结构的方式,认为像AlphaFold这样的程序基本上是一个黑盒子——它们提供了答案,但我们并不真正知道它们是如何做到的。
这个问题引起了哈萨比斯的兴趣,在诺贝尔奖宣布后的新闻发布会上,他指出:“我们目前正处于构建正确分析工具的早期阶段。”他想知道是否可以从神经科学中借鉴技术并将其应用于这一新问题。“对于AI系统来说,功能性磁共振成像扫描的等价物是什么——换句话说,一个虚拟大脑?”他问道。哈萨比斯还提出了未来或许可以建立类似AlphaFold这样的系统,使其能够用自然语言解释自身的工作原理,就像人类科学家一样。
在同一活动中,Jumper 表示他很谦卑,因为 AlphaFold 训练所用的每个数据点可能代表了世界各地某位博士生多年的工作。但是,尽管几代研究人员的努力使 AlphaFold 成为可能,Jumper 却惊叹于如今 AlphaFold 也能回报这份努力。“我几乎和此刻一样兴奋的是,在诺贝尔奖中提到使用 AlphaFold 理解某种疾病的研究工作——我迫不及待地期待这一刻。”
杰米·达兰尼是高级科学记者化学世界