
基于曼巴的材料基础模型
作者:Schmidt, Kristin
介绍
化学语言模型(LMS)的大规模培训方法(LMS)代表化学方面的显着进步1。这些方法在挑战分子任务(例如财产预测和分子产生)方面表现出了令人印象深刻的结果2。它们的成功在很大程度上归因于这些模型通过对广泛未标记的Corpora的自我监督学习来学习丰富的,上下文化的输入令牌的表述3。
当前可用的大多数化学基础模型都是基于变压器架构及其核心注意模块4,,,,5,,,,6。自我发挥的功效在于其在固定上下文窗口中密集路由信息的能力7,启用复杂化学数据的建模8。但是,这种机制固有地受到其无法在有限上下文窗口之外合并信息的限制,并且相对于序列长度,它遭受了二次缩放尺度。9。作为回应,大量研究致力于开发更有效的注意机制10。
结构化状态空间序列模型(SSM)最近已成为序列建模的有希望的替代方案11。这些模型结合了复发性神经网络(RNN)和卷积神经网络(CNNS)的特征12为了通过复发或卷积实现有效的计算,以序列长度提供线性或近线性缩放。Mamba是一种简化的端到端基于SSM的神经网络体系结构,既避开传统的关注,甚至是MLP块)13。Mamba提供快速的推断和与序列长度线性缩放,这对于处理化学数据集中遇到的经常长时间的微笑字符串尤其有利。
由于几个固有的优势,SSM特别适合对分子数据进行建模。首先,在处理广泛的化学数据库时,SSM的近线性缩放可以有效地处理长时间的微笑字符串14,其中大部分分子表现出长序列15。其次,复发和卷积元素的组合使SSM可以更有效地捕获局部化学相互作用(例如,键合模式)和全局结构依赖性(例如,总体分子拓扑),而不是固定膜片方法(例如变压器)16。这种结构细微差别的整体捕获是强大的分子性质预测,反应饲养估计和分子重建的基础。
在这项研究中,我们提出了一种新型的基于Mamba的大型基础模型,称为Osmi-SSM-336m。我们的编码器 - 模型基础模型经过有效的基于SSM的编码器,该模型与自动编码器机制对齐,该机制是在9100万个仔细策划分子的大型语料库上,可用于Pubchem17,导致40亿个分子令牌。我们的主要贡献如下:
-
我们预先训练了一个大规模的曼巴基分子基础模型,称为Osmi-SSM-336m,从Pubchem仔细策划的超过9100万个分子上17,相当于40亿个分子令牌。我们在11个基准数据集中进行了广泛的实验,其中包含量子机械,物理,生物物理和生理性能预测的小分子。
-
我们评估了使用Buchwald Hartwig交叉耦合反应数据集预测合成和过程化学场景中化学反应产量的能力,其中反应表示转化为产物的反应物的百分比。
-
我们在摩西基准测试数据集上评估模型的重建能力
-
18。我们提出了一项关于推理速度的比较研究,表明我们的基于MAMBA的模型优于基于变压器的模型,用于预测PubChem的1000万个随机选择样品的同性恋属性。
-
我们的结果表明O
smi-SSM-336m在各种任务中实现最新的性能,包括分子性质预测,反应饲养估计和分子重建。重要的是,所提出的模型在推理速度和预测性能之间取得了有效的平衡,提供最先进的模型会导致近一半的基于变压器的体系结构所需的时间。结果
实验
评估我们提出的基于曼巴的模型O的有效性
smi-SSM-336m,我们使用来自分子的11个数据集进行了实验19如表中所示1。具体而言,我们评估了六个数据集用于分类任务和五个用于回归任务的数据集。为了确保公正的评估,我们通过对所有任务采用相同的火车/验证/测试拆分来保持与原始基准的一致性19。我们还进行了实验考虑其他测试的十种不同种子,以确保该方法的鲁棒性。表1评估数据集说明
20。这些实验利用了三个1536孔板,覆盖了15个芳基和杂芳基卤化物的基质,四个布赫瓦尔德配体,三个碱基和23个依斯唑添加剂,总共产生了3955个反应。我们采用了与参考文献中相同的数据拆分。20用不同尺寸的训练集评估我们的模型性能。
评估OSMI-SSM-336的重建和解码器功能m,我们利用了摩西基准测试数据集18,其中包含1,936,962个分子结构。在实验中,我们采用了参考文献提出的数据集拆分。18,将其分为训练,测试和脚手架测试集,分别包括约160万,176,000和176,000个分子。脚手架测试集包括训练和测试集中缺乏的独特bemis-murcko脚手架,使我们能够评估模型生成先前未观察到的脚手架的能力。最后,我们评估了OSMI-SSM-336的推理速度m通过预测从PubChem随机选择的1000万样品的同性恋性质。与SOTA在基准测试任务中进行比较
分类任务的结果
分析评估了O的比较性能
smi-SSM-336m如表中所述2。表2 Moleculenet基准数据集的分类任务的方法和性能桌子
总结了用于分子分类任务的几个基准测试数据集中各种高级方法的性能。OSMI-SSM-336m证明了针对基于变压器的方法的比较功效,在六个数据集中的三个中表现出色。值得注意的是,OSMI-SSM-336m其初始配置与当前最新方法相当。O的进一步微调Osmi-SSM-336m提高其性能,表明其具有准确分子分类的巨大潜力,并表明可以通过进一步优化实现额外的性能提高21,,,,22,,,,23,,,,24,,,,25。回归任务的结果随后,我们应用了O
smi
-SSM-336m预测化学性质。表格列出3。表3分子基准数据集回归任务的方法和性能表中显示的结果
表示OSMI-SSM-336m实现与最新模型相当的性能,在评估的五个回归基准中,获得了第二好的结果。这证明了基于MAMBA的方法在与基于变压器的方法相当的情况下提供结果的功效,同时也突出了其在一系列化学性质预测任务中的鲁棒性。OSMI-SSM-336的设计m旨在在预测准确性和推理效率之间取得最佳平衡。为了举例说明这一平衡,我们提供了一个分析,比较了在数据集中预测从PubChem随机选择的1000万个样本的数据集上的推理时间。这项研究强调了模型保持高预测准确性的能力,同时大大减少了计算时间,从而为大型化学性质预测提供了实际优势。
Humo-Lumo属性预测的速度推断
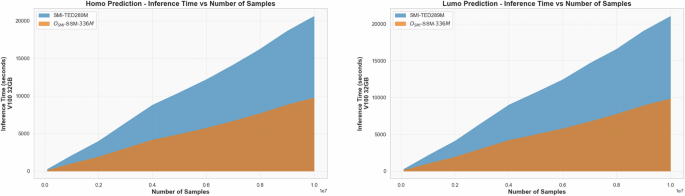
为了评估所提出的基于MAMBA的方法的推理速度,我们对从PubChem随机选择的1000万样品进行了对同性恋性质的预测。为了进行比较,我们评估了SMI-TED289M的推理时间,这是一种基于变压器的模型,以其最先进的性能认可。数字1说明OSMI-SSM-336的卓越推理速度m与SMI-TED89M相比。具体而言,使用单个NVIDIA V100 V100 32GB GPU,SMI-TED289M需要20,606.76来进行HOMO性质预测和21,038.43的21,038.43。相反,OSMI-SSM-336m在同一GPU上的9823.64 s中完成了9735.64 s和lumo预测的HOMO预测。这些结果突出了O的大量效率提高smi-SSM-336m在推理速度方面的模型。图1

m考虑到从PubChem和单个NVIDIA V100 32GB GPU中随机选择的10M样品的数据集,用于HOMO-LUMO预测的SMI-TED289M。MAMBA-BASE方法表明效率有很大的提高,大约54%的速度,将GPU的使用量减少了6 H,同时也将CO2排放量平均降低0.78 kg等效。
26。计算资源的减少对于最大程度地降低机器学习模型的环境影响至关重要,这需要大量的能耗和相关的碳足迹27。反应收益预测以前,我们能够证明,与单分子性能预测相比,提出的基于MAMBA的模型能够执行与基于变压器的方法相比。
在这里,我们研究了基于MAMBA的化学反应方法。
通过编写由箭头隔开的反应物和产物的结构公式来描述有机化学中的化学反应,通过指定一个或几个反应剂分子和一个或几个产物分子之间的原子重新排列来代表化学转化。预测化学反应的结果,例如基于在高通量筛选中收集的数据的产量,是化学的机器学习的重要任务。数字2显示了化学反应的模式。

该图说明了考虑O的反应微笑的化学反应饲养预测的模式smi-SSM-336m模型。我们使用Buchwald Hartwig交叉偶联反应的高通量数据集评估了这种结构,以预测反应产量20。
这涉及估计转换为产品的反应物的百分比。我们的评估遵循参考文献中概述的模式和数据划分。20。桌子4呈现O的结果smi-SSM-336m模型并将其性能与现有的最新方法进行比较。表4 O的性能smi
清楚地证明了针对最先进方法的基准测试时,提出的基于MAMBA的基础模型的优势,包括梯度增强和基于指纹的方法(DRFP)28,基于DFT的随机森林模型(DFT)28,以及基于变形金刚的模型,例如ford-bert29以及它的增强变体,产量 - 伯特(8月)29和MSR2-RXN30。基于MAMBA的模型的性能可以归因于其在9100万个策划分子的广泛数据集中的预培训,该数据集提供了强大的化学知识基础,从而显着增强了其预测能力。这种预训练使该模型即使使用有限的培训数据也能够达到高精度,这是在仅2.5%的可用样品中训练的情况下的持续性能来证明的。为了确保模型的鲁棒性,我们用十个不同的随机种子进行了每个实验。
一个关键的观察结果是在各种数据拆分中的模型鲁棒性,尤其是在低资源设置中,只有一小部分数据集用于培训。这种弹性强调了利用大规模预训练对广义化学知识进行编码的重要性,然后可以对这些知识进行微调,以用于反应收益预测等特定任务。相反,专门针对给定任务量身定制的模型倾向于过分地适合培训数据的细微差别,并在何时降低训练设置时努力概括,从而突出了其设计的关键限制。
此外,基于MAMBA的模型的鲁棒性扩展到其在室外测试集上的性能。概括与训练集不同的数据分布的能力是模型评估的关键方面,尤其是在化学反应多样性的现实应用程序中。基于Mamba的模型在内域和室外测试集中的一致性表明,预训练对多样化且全面的数据集的功效,这使该模型具有处理广泛的化学环境和反应条件的灵活性。
基于MAMBA的模型与其他最先进方法之间的比较分析还阐明了传统方法(例如基于DFT)模型的局限性,尽管在量子化学方面,它们在理论上基础,但在实际情况下可能无法捕获反应机制的全部复杂性。同样,尽管基于变压器的模型(例如产量 - 伯特及其增强变体表现出强大的性能),但它们却没有基于曼巴的模型,尤其是在低数据表方面,表明训练前数据的纯粹规模和多样性在实现出色的结果方面起着至关重要的作用。
这些发现强调了化学基础模型的潜力,在化学中,在大型,多样的数据集中进行预培训可以作为开发不仅准确而且可以稳健且可推广的模型的强大范式。这项工作的含义超出了反应收益的预测,这表明可以将类似的策略应用于计算化学和材料科学中的其他领域,在计算化学和材料科学中,跨不同数据集的能力至关重要。
对摩西基准测试数据集的解码器评估
接下来,对O进行了比较评估smi-SSM-336m使用包含176,000个分子的测试集,针对微笑重建和解码的几种基线模型进行了模型。评估指标,表中详细列出5,在关键领域(例如碎片相似性(Frag),脚手架相似性(SCAF),与最近的邻居(SNN),内部多样性(INTDIV)和FRâ©CHET CHEMNET距离(FCD)等相似性,提供了模型的全面视图。
结果表明Osmi-SSM-336m不仅匹配,而且超过了最先进的模型在产生独特,有效和新颖的分子方面的性能。它在碎片度量中的近乎完美的评分突出了其保持分子片段结构完整性的显着能力,这是确保产生的分子在化学上保持可行性且与现实世界应用相关的关键方面。这种高片段相似性,再加上模型的低FCD评分,这表明产生的分子的分布紧密反映了天然分子的分布。
除了碎片级别的精度外,osmi-SSM-336m在支架相似性(SCAF)和最近的邻居相似性(SNN)中表现出卓越的性能。这些指标在药物发现和设计中尤为重要,在药物发现和设计中,核心分子支架的保存对于维持生物学活性至关重要。该模型的生成具有高支架相似性的分子的能力表明,它可以可靠地再现分子的核心结构特征,这是生成保留其预期生物学功能的候选化合物的要求。
另一个重要的发现是内部多样性(INTDIV)的模型表现。尽管高相似性得分很重要,但生成的集合中的多样性同样至关重要,尤其是在需要对化学空间进行广泛探索的情况下。osmi-SSM-336m模型实现了值得称赞的平衡,保持高相似性指标,同时还产生具有实质成对差异的分子。这种能力产生各种分子而不牺牲结构完整性的能力使该模型对于在药物发现中的应用高度有价值,在这种模型中,通常需要探索广泛的化学可能性来识别最佳候选者。
此外,与传统方法(例如Charrnn)以及JT-VAE和Molgen-7B(OSMI-SSM-336)等传统方法相比m模型在所有评估的指标上始终胜过表现。这包括诸如豪华轿车之类的模型,尽管它具有强大的内部多样性,但仍无法与其他指标相匹配,这表明在这些方法中进行了权衡,即OSMI-SSM-336m成功减轻。该模型的实现高支架相似性的能力同时保持多种分子结构表明,其在大规模数据集上的预训练使其对化学空间具有广泛的理解,从而使其能够在各种分子构构中有效地概括。
讨论
本文介绍Osmi-SSM-336m这是一种基于MAMBA的化学基础模型,该模型在Pubchem的9100万个微笑样本的策划数据集上进行了预先训练,其中包括40亿个分子令牌。该模型旨在在评估指标的高性能和更快的推理功能之间达到平衡。
O的功效smi-SSM-336m在各种任务中进行了严格评估,包括分子财产分类和预测。该模型不仅取得了最新的结果,而且还显示出显着提高效率。具体而言,它比现有的基于最新变压器的方法快约54%,将GPU的使用降低6 H,并将CO2排放降低为0.78 kg CO2同等含量。26在预测1000万个从PubChem中随机选择样品的数据集的Homo-Lumo间隙期间。
我们还探讨了模型在预测化学反应结果方面的功能,例如基于高通量筛选数据的反应产量,这是化学机器学习的关键任务。基于MAMBA的模型在内域和室外测试集中的一致性凸显了预训练对多样化和全面数据集的有效性。这种预训练使该模型能够适应广泛的化学环境和反应条件。我们的比较分析表明,尽管基于DFT的模型等传统方法基于量子化学,但它们可能无法完全捕获实际情况下反应机制的复杂性。同样,基于变压器的模型,例如产量 - 伯特及其增强变体,尽管其性能很强,但基于MAMBA的模型表现出色,尤其是在低数据制度中。这突出了大规模,多样化的预训练数据在取得卓越结果中发挥的关键作用。
最后,我们对O的O进行了比较评估smi-SSM-336m针对微笑重建和解码的几个基线模型的模型。模型的各种指标的性能表明了利用大规模数据集进行预训练的重要性,这可能导致模型不仅在产生高质量的分子方面表现出色,而且还具有应对计算化学和药物设计中的复杂挑战所需的灵活性。
本文介绍的基于MAMBA的基础模型为广泛的科学应用提供了灵活性和可扩展性。
方法
本节概述了拟议的Osmi-SSM-336m小分子的基础模型。在这里,我们概述了收集,策展和预处理培训数据的过程。此外,我们描述了令牌编码器过程和笑容编码器过程。
预训练数据
预培训数据来自PubChem数据存储库,该数据库包含有关化学物质及其生物活动的信息17。最初,从Pubchem收集了1.13亿个微笑弦。这些分子字符串使用RDKIT中实施的标准程序进行了重复数据删除和规范化。31,,,,32;每个微笑串都标准化,转换为规范形式,并通过检查一致的价和粘合规则来进行消毒,以确保唯一性和化学有效性。此后,应用了一个分子转化过程,并结合了额外的化学消毒检查,以验证从独特的微笑字符串中得出的分子,从而导致最终策划的9100万个独特和化学声音分子。
为了构建词汇,我们利用了参考文献提出的分子令牌。33。令牌化过程应用于Pubchem的所有9100万个策划分子,产生40亿个分子令牌。通过此输出,我们提取了2988个独特的令牌,以及5个特殊令牌。相比之下,对以最小策划进行了10亿个样品训练的Moleformer,使用相同的令牌化方法产生了2362个令牌的词汇2。这表明我们的全面策划过程导致了增强和更具代表性的词汇模型。表中提供了预训练数据集的详细统计数据6。
模型架构
我们为O进行培训smi-SSM-336m使用基于MAMBA的代币编码器以及编码器编码器体系结构有效地将Smiles序列映射到潜在的嵌入式空间和背部。osmi-SSM-336m设计利用SSM的优势来捕获远程依赖性和近线性缩放的过程序列。在我们的方法中,编码器过程通过mamba块输入令牌,而解码器则重建这些嵌入以准确地生成微笑序列。表中指定了模型的超参数
7。隐藏尺寸设置为768,总计24层。重要的是要强调,与具有相似大小的变压器相比,我们基于MAMBA的模型中的层计数有效地翻了一番。在传统的变压器中,每一层包括多头注意(MHA)块,然后是多层感知器(MLP)块。相反,我们的体系结构为每个这样的变压器层实现了两个不同的MAMBA块,一个处理类似于MHA的功能,而另一个与MLP相对应的函数,导致层数增加了一倍。
参数DT最小值(0.001),DT MAX(0.1),DT量表(1.0)和DT Init Floor(1eâ4)控制我们模型基础的连续时间系统的动力学,确保稳定的训练和有效的表示学习。
MAMBA模型源自映射输入函数或序列的连续时间系统\(x(t)\ in {{\ mathbb {r}}}}^{m} \)输出响应信号\(y(t)\,\ in \,{{\ mathbb {r}}}}}^{o} \)\)通过隐式潜在状态\(h(t)\,\ in \,{{\ mathbb {r}}}}}^{n} \)\)可以使用以下普通差分等式在数学上进行数学配方。((1):
$ \ begin {array} {rcl} {h}^{{{\ prime}}(t)&=&ah(t)+bx(t),\\ y(t)&=&=&=&ch(t)+dx(t)+dx(t)
(1)
在哪里\(a \,\ in \,{{\ Mathbb {r}}}}}^{n \ times n} \)和\(c \,\ in \,{{\ Mathbb {r}}}}}^{o \ times n} \)控制当前状态如何随着时间的流逝而演变,并转化为输出,\(b \,\ in \,{{\ Mathbb {r}}}}}^{n \ times m} \)和\(d \,\ in \,{{\ Mathbb {r}}}}}^{o \ times m} \)描述输入如何分别影响状态和产出。
从微笑槽中提取的令牌SSM编码器嵌入在768维空间中。编码器层层旨在处理分子令牌嵌入,表示为\({\ bf {x}}} \,\ in {{\ mathbb {r}}}}}}^{t \ times l} \), 在哪里t表示令牌的最大数量和l表示嵌入空间维度。我们有限t在202令牌时,Pubchem数据集中的99.4%的分子包含的令牌少于此阈值2。在仅编码模型中,通常使用平均合并层表示代币作为潜在空间中的微笑34。
但是,这种方法受到平均合并操作缺乏自然反转过程的限制。为了克服这一限制,我们旨在通过累积的x在潜在空间中,称为z,如等式中所述。((2):
$$ {\ bf {z}} = \ left(\,\ text {layernorm} \,\ left(\,\,\ text {gelu} \,\,\ left({\ bf {{\ bf {x}} {{\ bf {w}}} _ {1}+{{{\ bf {bf {b}}} _ {1} \ right)\ right)\ right)\ right)\ right){{\ bf {w}}}}}} _ {2} _ {2},$ {2},$ $
(2)
在哪里\({\ bf {z}}} \ in {{\ mathbb {r}}}}}}}^{l} \)\),,,,\({{{\ bf {w}}} _ {1} \ in {{\ Mathbb {r}}}}}^{l} {l} \),,,,\(({{\ bf {b}}}} _ {1} \ in {{\ Mathbb {r}}}}}^{l} {l} \),,,,\({{{\ bf {w}}} _ {2} \ in {{\ Mathbb {r}}}}}^{l \ times l} \), 和l表示潜在空间尺寸(特别是l= 768)。随后,我们可以沉浸z通过计算等式返回。((3):
$ \ hat {{\ bf {x}}}} = \ left(\,\ text {layernorm} \,\ left(\,\ text {gelu} \,bf {z}} {{\ bf {w}}} _ {3}+{{{\ bf {bf {b}}} _ {3} \ right)\ right)\ right)\ right)\ right)
(3)
在哪里\(\ hat {{\ bf {x}}}} \ in {{\ mathbb {r}}}}}}^{t \ times l} \),,,,\({{{\ bf {w}}} _ {3} \ in {{\ mathbb {r}}}}}^{l \ times l} \),,,,\({{{\ bf {b}}} _ {3} \ in {{\ mathbb {r}}}}}^{l} \),,,,\({{{\ bf {w}}} _ {4} \ in {{\ mathbb {r}}}}}^{l \ times t} \)。在哪里t表示输出特征空间大小(即t= 202)。
语言层(解码器)用于处理\(\ hat {{\ bf {x}}}} \),在应用非线性和归一化的情况35。该体系结构是分子结构领域中降低和表示学习的工具。
训练前策略
O的预训练smi-SSM-336mwas performed for 130 epochs on the entire curated PubChem dataset using a fixed learning rate of 3 × 10−5and a batch size of 128 molecules on 24 NVIDIA V100 (16G) GPUs, distributed across 4 nodes via DDP andtorch run。The pre-training process is divided into two distinct phases:
-
Phase 1: in this initial phase, the token encoder is pre-trained using 95% of the available samples, while the remaining 5% is reserved exclusively for training the encoder-decoder layer.This partitioning is necessary to mitigate convergence difficulties in the early epochs of token embedding learning, which could otherwise adversely affect the training of the encoder-decoder component.
-
Phase 2: once the token embeddings have converged, the pre-training is expanded to utilize 100% of the available samples for both phases.This approach enhances the performance of the encoder-decoder layer, particularly in terms of token reconstruction accuracy.
For encoder pre-training, we employ the masked language modeling strategy as defined in ref.36。Initially, 15% of the tokens are selected for possible prediction;of these, 80% are replaced with the [MASK] token, 10% are substituted with a random token, and the remaining 10% remain unchanged.This strategy facilitates the learning of robust, contextualized representations of the SMILES tokens.
Our approach also incorporates a latent space embedding mechanism that supersedes conventional mean pooling.Instead of aggregating token embeddings via an averaging operation (which lacks a natural inversion process), the token embeddings are transformed into a latent vectorz(as detailed in Eqs. (2) 和 (3) of the “Model architecture†section).This latent representation captures intricate structural nuances of the SMILES strings and supports a reversible mapping, thereby enabling both accurate SMILES reconstruction and effective downstream tasks such as molecular property prediction.
The pre-training procedure is guided by two distinct loss functions.The first loss function, based on the masked language model objective, uses cross-entropy loss to predict the masked tokens.The second loss function governs the reconstruction task and is measured using the mean squared error (MSE) between the original tokens and their reconstructions generated by the encoder-decoder layer.Monitoring these metrics ensures the convergence of the token embeddings and the stability of the latent space representation throughout training.
数据可用性
No datasets were generated or analyzed during the current study.
代码可用性
All Python codes for training and fine-tuning Osmi-SSM-336m, together with Python notebooks for experimental evaluations, are available athttps://github.com/IBM/materials/tree/main/models/smi_ssed。Pre-trained model weights can be accessed via our HuggingFace repository athttps://huggingface.co/ibm-research/materials.smi_ssed。For other enquiries contact the corresponding authors.
参考
Sadybekov, A. V. & Katritch, V. Computational approaches streamlining drug discovery.自然 616, 673–685 (2023).
文章一个 Google Scholar一个
Ross, J. et al.Large-scale chemical language representations capture molecular structure and properties.纳特。马赫。Intell。 4, 1256–1264 (2022).
文章一个 Google Scholar一个
Bommasani, R. et al.On the opportunities and risks of foundation models.Preprint atarxiv https://doi.org/10.48550/arXiv.2108.07258(2021).
Pesciullesi, G., Schwaller, P., Laino, T. & Reymond, J.-L.Transfer learning enables the molecular transformer to predict regio-and stereoselective reactions on carbohydrates.纳特。社区。 11, 4874 (2020).
文章一个 Google Scholar一个
Chithrananda, S., Grand, G. & Ramsundar, B. Chemberta: large-scale self-supervised pretraining for molecular property prediction.Preprint atarxiv https://doi.org/10.48550/arXiv.2010.09885(2020)。
Janakarajan, N., Erdmann, T., Swaminathan, S., Laino, T. & Born, J. Language models in molecular discovery.Preprint atarxiv https://doi.org/10.48550/arXiv.2309.16235(2023)。
Vaswani, A. et al.注意就是您所需要的。在神经信息处理系统的进步30 (2017).
Tay, Y., Dehghani, M., Bahri, D. & Metzler, D. Efficient transformers: a survey.ACM计算。幸存。 55, 1–28 (2022).
文章一个 Google Scholar一个
Lin, T., Wang, Y., Liu, X. & Qiu, X. A survey of transformers.AI Open 3, 111–132 (2022).
文章一个 Google Scholar一个
Kotei, E. & Thirunavukarasu, R. A systematic review of transformer-based pre-trained language models through self-supervised learning.信息 14, 187 (2023).
文章一个 Google Scholar一个
Gu, A., Goel, K. & Ré, C. Efficiently modeling long sequences with structured state spaces.Preprint atarxiv https://doi.org/10.48550/arXiv.2111.00396(2021).
Smith, J. T., Warrington, A. & Linderman, S. W. Simplified state space layers for sequence modeling.Preprint atarxiv https://doi.org/10.48550/arXiv.2208.04933(2022)。
Gu, A. & Dao, T. Mamba: Linear-time sequence modeling with selective state spaces.Preprint atarxiv https://doi.org/10.48550/arXiv.2312.00752(2023)。
Patro, B. N. & Agneeswaran, V. S. Mamba-360: Survey of state space models as transformer alternative for long sequence modelling: Methods, applications, and challenges.Preprint atarxiv https://doi.org/10.48550/arXiv.2404.16112(2024)。
Hähnke, V. D., Kim, S. & Bolton, E. E. Pubchem chemical structure standardization.J. Cheminformatics 10, 1–40 (2018).
文章一个 Google Scholar一个
Waleffe, R. et al.An empirical study of mamba-based language models.Preprint atarxiv https://doi.org/10.48550/arXiv.2406.07887(2024)。
Kim, S. et al.Pubchem 2023 update.核酸res。 51, D1373–D1380 (2023).
文章一个 Google Scholar一个
Polykovskiy, D. et al.Molecular sets (moses): a benchmarking platform for molecular generation models.正面。Pharmacol。 11, 565644 (2020).
文章一个 Google Scholar一个
Wu,Z。等。Moleculenet: a benchmark for molecular machine learning.化学科学。 9, 513–530 (2018).
文章一个 Google Scholar一个
Ahneman, D. T., Estrada, J. G., Lin, S., Dreher, S. D. & Doyle, A. G. Predicting reaction performance in c–n cross-coupling using machine learning.科学 360, 186–190 (2018).
文章一个 Google Scholar一个
Yang, K. et al.Analyzing learned molecular representations for property prediction.J. Chem。inf。模型。 59, 3370–3388 (2019).
文章一个 Google Scholar一个
Liu, S., Demirel, M. F. & Liang, Y. N-gram graph: simple unsupervised representation for graphs, with applications to molecules.在神经信息处理系统的进步32 (2019).
Hu, W. et al.Strategies for pre-training graph neural networks.Preprint atarxiv https://doi.org/10.48550/arXiv.1905.12265(2019)。
Wang, Y., Wang, J., Cao, Z. & Barati Farimani, A. Molecular contrastive learning of representations via graph neural networks.纳特。马赫。Intell。 4, 279–287 (2022).
文章一个 Google Scholar一个
Hu, Z., Dong, Y., Wang, K., Chang, K.-W.& Sun, Y. Gpt-gnn: generative pre-training of graph neural networks.在Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, 1857–1867 (2020).
Lacoste, A., Luccioni, A., Schmidt, V. & Dandres, T. Quantifying the carbon emissions of machine learning.Preprint atarxiv https://doi.org/10.48550/arXiv.1910.09700(2019)。
Rillig, M. C., Ã…gerstrand, M., Bi, M., Gould, K. A. & Sauerland, U. Risks and benefits of large language models for the environment.环境。科学。技术。 57, 3464–3466 (2023).
文章一个 Google Scholar一个
Probst, D., Schwaller, P. & Reymond, J.-L.Reaction classification and yield prediction using the differential reaction fingerprint drfp.Digital Discov. 1, 91–97 (2022).
文章一个 Google Scholar一个
Schwaller, P., Vaucher, A. C., Laino, T. & Reymond, J.-L.Prediction of chemical reaction yields using deep learning.马赫。学习。科学。技术。 2, 015016 (2021).
文章一个 Google Scholar一个
Boulougouri, M., Vandergheynst, P. & Probst, D. Molecular set representation learning.纳特。马赫。Intell。 6, 754–763 (2024).
Landrum, G. Rdkit documentation.发布 1, 4 (2013).
Heid, E., Liu, J., Aude, A. & Green, W. H. Influence of template size, canonicalization, and exclusivity for retrosynthesis and reaction prediction applications.J. Chem。inf。模型。 62, 16–26 (2021).
文章一个 Google Scholar一个
Schwaller, P. et al.Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction.ACS Cent.科学。 5, 1572–1583 (2019).
文章一个 Google Scholar一个
Bran, A. M. & Schwaller, P. Transformers and large language models for chemistry and drug discovery.Preprint atarxiv https://doi.org/10.48550/arXiv.2310.06083(2023)。
Ferrando, J., Gállego, G. I., Tsiamas, I. & Costa-jussà , M. R. Explaining how transformers use context to build predictions.Preprint atarxiv https://doi.org/10.48550/arXiv.2305.12535(2023)。
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. Bert: pre-training of deep bidirectional transformers for language understanding.在North American Chapter of the Association for Computational Linguistics(2019)。
Liu,S。等。Pre-training molecular graph representation with 3d geometry.Preprint atarxiv https://doi.org/10.48550/arXiv.2110.07728(2021).
Fang, X. et al.Geometry-enhanced molecular representation learning for property prediction.纳特。马赫。Intell。 4, 127–134 (2022).
文章一个 Google Scholar一个
Rong, Y. et al.Self-supervised graph transformer on large-scale molecular data.ADV。神经信息。过程。系统。 33, 12559–12571 (2020).
Ahmad, W., Simon, E., Chithrananda, S., Grand, G. & Ramsundar, B. Chemberta-2: towards chemical foundation models.Preprint atarxiv https://doi.org/10.48550/arXiv.2209.01712(2022)。
Taylor, R. et al.Galactica: A large language model for science.Preprint atarxiv https://doi.org/10.48550/arXiv.2211.09085(2022)。
Zhou, G. et al.Uni-mol: a universal 3d molecular representation learning framework.ChemRxiv(2023)。
Chang, J. & Ye, J. C. Bidirectional generation of structure and properties through a single molecular foundation model.纳特。社区。 15, 2323 (2024).
文章一个 Google Scholar一个
Soares, E. et al.A large encoder-decoder family of foundation models for chemical language.Preprint atarxiv https://doi.org/10.48550/arXiv.2407.20267(2024)。
Jin, W., Barzilay, R. & Jaakkola, T. Junction tree variational autoencoder for molecular graph generation.在International Conference on Machine Learning, 2323–2332 (PMLR, 2018).
Eckmann, P. et al.Limo: latent inceptionism for targeted molecule generation.Proc。马赫。学习。res。 162, 5777 (2022).
Fang, Y. et al.Domain-agnostic molecular generation with self-feedback.Preprint atarxiv https://doi.org/10.48550/arXiv.2301.11259(2023)。
Ross, J. et al.Gp-molformer: a foundation model for molecular generation.Preprint atarxiv https://doi.org/10.48550/arXiv.2405.04912(2024)。
道德声明
竞争利益
作者没有宣称没有竞争利益。
附加信息
Publisher’s noteSpringer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
权利和权限
开放访问This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material.您没有根据本许可证的许可来共享本文或部分内容的改编材料。The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visithttp://creativecommons.org/licenses/by-nc-nd/4.0/。重印和权限
引用本文

Soares, E., Vital Brazil, E., Shirasuna, V.
等。A Mamba-based foundation model for materials.npj Artif.Intell。 1, 8 (2025).https://doi.org/10.1038/s44387-025-00009-7
已收到:
公认:
出版:
doi:https://doi.org/10.1038/s44387-025-00009-7