
Target sequence-conditioned design of peptide binders using masked language modeling
作者:Chatterjee, Pranam
Main
The development of therapeutics largely relies on the ability to design small-molecule-based or protein-based binders to pathogenic target proteins of interest1. These binders can be used either as inhibitors or as functional recruiters of effector enzymes2. For example, proteolysis-targeting chimeras (PROTACs) or molecular glues are heterobifunctional small molecules that bind and recruit endogenous E3 ubiquitin ligases for targeted protein degradation (TPD)3,4. Still, these small-molecule-based methods rely on the existence of accessible cryptic or canonical binding sites, which are not present on classically ‘undruggable’ intracellular proteins5,6. With the advent of deep-learning-based structure prediction tools such as AlphaFold2 and AlphaFold3 (refs. 7,8), combined with generative modeling1, algorithms such as RFdiffusion and MASIF-Seed enable researchers to conduct de novo protein binder design from target structure alone9,10. Nonetheless, much of the undruggable proteome, including dysregulated proteins such as transcription factors and fusion oncoproteins, are conformationally disordered, thus biasing design to a small subset of disease-related proteins1,6.
Over the past few years, deep learning has revolutionized natural language processing (NLP), particularly through the implementation of the attention mechanism11. This foundational advancement has transcended the boundaries of natural language analysis, finding applications in the modeling of other languages, such as proteins, which are fundamentally sequences of amino acids12. Recently, several protein language models (pLMs) trained on distinct transformer architectures, such as ProtT5, ProGen2, ProtGPT2 and the ESM series, have accurately captured critical physicochemical properties of proteins13,14,15,16. Notably, ESM-2 currently stands as a state-of-the-art model in the realm of protein sequence representation, essentially functioning as an encoder-only model that discerns co-evolutionary patterns among protein sequences via a masked language modeling (MLM) training task17,18. These models have been extended to powerful applications, including antibody design, the creation of novel proteins and structure prediction, offering a streamlined approach to embedding useful protein information14,15,17,18. Recently, our laboratory has leveraged the expressivity of pLMs to both generate and prioritize effective peptidic binder motifs to targets of interest, enabling design of peptide-guided protein degraders19,20 that are modeled after the ubiquibody (uAb) architecture developed by Portnoff et al.21,22. As such, uAbs now represent a programmable, CRISPR-like approach for TPD. Our early models, Cut&CLIP and SaLT&PepPr, rely on the existence of interacting partner sequences as scaffolds for peptide design19,23. Most recently, our PepPrCLIP model generates de novo peptides by first sampling the ESM-2 latent space for naturalistic peptide candidates and then screening these candidates through a contrastive model to determine target sequence specificity20. However, a purely de novo, target sequence-conditioned binder design algorithm has yet to be developed.
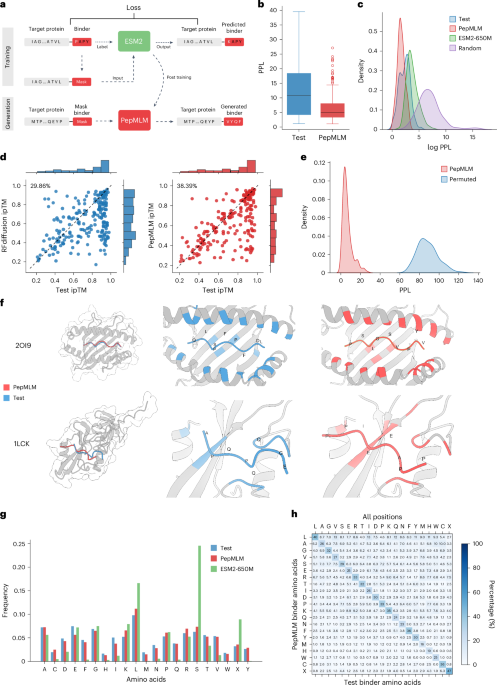
To achieve this goal, we introduce PepMLM, a Peptide binder design algorithm via Masked Language Modeling, built upon the foundations of ESM-2 (ref. 17). PepMLM employs a masking strategy that uniquely positions the entire peptide binder sequence at the terminus of target protein sequences, compelling ESM-2 to reconstruct the entire binding region (Fig. 1a). PepMLM-derived linear peptides achieve low perplexities, matching or improving upon validated peptide–protein sequence pairs in the test dataset; outperform the state-of-the-art RFdiffusion model for peptide design on structured targets in silico9; and experimentally exhibit potent and specific binding to disease-relevant targets and degradation of difficult-to-drug drivers of Huntington’s disease and emergent viral phosphoproteins when incorporated into the uAb architecture. Overall, by focusing on the complete reconstruction of peptide regions, PepMLM serves as a completely sequence-based, target-conditioned de novo binder design tool, paving the way for the development of more effective, therapeutic binders to conformationally diverse proteins of interest.

a, The architecture of the PepMLM model. Based on the finetuning of ESM-2, the model incorporates the target protein sequence along with a masked binder region during the training phase. During the generation phase, the model can accept target protein sequences and mask tokens to facilitate the creation of peptides of specified lengths. b, Perplexity distribution comparison. The perplexity values were calculated for test and designed peptides, encompassing the target proteins in the test set. c, The density distribution visualization of the log perplexity values for target–peptide pairs, encompassing test peptides, PepMLM-650M-designed peptides, ESM-2-650M-designed peptides and random peptides. d, In silico hit rate assessment of RFdiffusion (left) and PepMLM (right). Using AlphaFold-Multimer, ipTM scores were computed for both the designed and test peptides in conjunction with the target protein sequence. The entries are organized in accordance with the ipTM scores attributed to the test set peptides. The hit rate is characterized by the designed peptides exhibiting ipTM scores ≥ those of the test peptides. e, Binding specificity analysis through permutation tests. The distribution of PPL scores for matched target–binder pairs (blue) is compared with randomly shuffled mismatched pairs (red). Each target’s binder was shuffled 100 times to generate the mismatched distribution. Statistical significance was determined using t-test (P < 0.001). f, Structural comparison of computationally designed and experimental peptide binders in complex with their target proteins. Target proteins (gray) are shown in complex with PepMLM-designed binders (red) and experimental test binders (blue), with contact residues highlighted in corresponding colors. Top, mouse H-2Kb MHC complex (PDB ID: 2OI9) with designed peptide PSLGSVPYV (ipTM: 0.9) and test peptide QLSPFPFDL (ipTM: 0.9). Bottom, human tyrosine kinase complex (PDB ID: 1LCK) with designed peptide PPAEEIPP (ipTM: 0.82) and test peptide EGQQPQPA (ipTM: 0.68). g, Frequency distribution of individual amino acids among peptide binders (n = 203), comparing the test set (blue), PepMLM-designed sequences (red) and ESM2-650M-designed sequences (green). h, Amino-acid-specific generation distribution at contact positions (8-Å threshold). The heatmap shows the percentage of designed amino acids (y axis) given each amino acid in test binders (x axis).
Results
PepMLM leverages span masking via ESM-2 embeddings for de novo design of target-binding peptides
We trained PepMLM using existing peptide–protein binding data sourced from the recent PepNN training set and the gold standard Propedia dataset24,25. We subjected our curated dataset to a filtration process based on the lengths of the binder and target protein sequences, which were confined to 50 and 500, respectively. To remove redundancies, we applied MMSeq2 clustering with a threshold at 80% on target protein sequences and removed entries that are in the same cluster and have the same binder sequence. The final dataset was split into 10,000 samples for training and 203 samples for testing26. Each entry in the dataset comprised a concatenated protein and binder sequence. During the training phase, we masked the entire peptide sequence, tasking the model to reconstruct them via the ESM-2-650M model17. The discrepancy between the ground truth binder and the reconstructed binder induces a cross-entropy loss, thereby forcing parameter updates via gradient descent. After finetuning, we generate peptide binders of specific lengths by providing the model with a target protein sequence and a user-defined number of mask tokens, as illustrated in Fig. 1a. Final settings and hyperparameters used to train our model are presented in Supplementary Table 1. We used pseudo-perplexity (PPL) of PepMLM to score binders given target protein sequence (Methods). Two decoding strategies were implemented: a default greedy decoding approach17 that selects the highest probability token at each site and a top-k sampling method that chooses from among the k most probable tokens to generate diverse peptide binders. After testing various k values (2–20) and analyzing the tradeoff between sequence diversity and model confidence through perplexity measurements, we selected k = 3 as the optimal value for balancing these factors (Supplementary Fig. 1).
To substantiate the efficacy of the designed peptides, we conducted a comprehensive series of computational benchmarks with test set peptide–target pairs. The total 203 test set target proteins were used to generate one peptide binder each in one shot, employing pretrained ESM-2 embeddings and PepMLM with additional randomly designed binders. All three groups (PepMLM, ESM-2 and Random) use the same length as the ground truth test binder for each target protein. Subsequently, the PPL of the binder region was computed for four groups of target protein–binder pairs. For a majority of the test set, known binders exhibited a reasonable perplexity range, with only a few outliers (those with a perplexity >40), validating the effective ability of PepMLM to model them accurately (Fig. 1b and Supplementary Table 2). Our distribution analysis revealed that PepMLM closely mirrors the low PPL region of real binders, a deviation from the distribution shifts observed with the original ESM-2 model alone and with randomly designed binders, indicating that PepMLM can distinguish binders from non-binders especially for random binders by PPL scores (Fig. 1c).
To further understand PPL score, we co-folded the test binders with their respective target proteins using AlphaFold-Multimer, which has been proven effective at predicting peptide–protein complexes27,28. The predicted local distance difference test (pLDDT) and interface-predicted template modeling (ipTM) scores, verified metrics within AlphaFold2 (ref. 7), function as critical indicators of the structural integrity and the potential interface binding affinity of the peptide–protein complex, respectively, providing an external quantitative assessment of our generation. PPL, our confidence metric, showed significant agreement with folding scores, as the extracted ipTM and pLDDT values from our benchmarking indicated a statistically significant negative correlation (P < 0.01) with PepMLM PPL. This demonstrates that our model can reflect binding interactions in a zero-shot manner via PPL while validating its reliability in prioritizing stable target-binding molecules (Supplementary Fig. 2).
To evaluate our generation quality, we compared PepMLM with RFdiffusion by generating one binder per target and performing structural prediction using AlphaFold-Multimer. We used the ipTM scores of test binders as reference points, defining a successful hit when a designed binder achieved a higher ipTM score than its corresponding test binder, indicating AlphaFold’s prediction of stronger binding affinity. Our analysis revealed hit rates of 38% for PepMLM and 29% for RFdiffusion (Fig. 1d). When applying more stringent criteria with plDDT scores greater than 0.8, PepMLM and RFdiffusion achieved hit rates of 49% and 34%, respectively (Supplementary Fig. 3). These results demonstrate the superior performance of PepMLM in peptide binder design, even for structured targets, potentially reducing the need for extensive experimental screening.
To investigate binding specificity of designed binders, we conducted a permutation test across 203 target–binder pairs using the PPL metric. For each target, we performed 100 random binder shuffles and computed PPL scores for these mismatched pairs. Statistical analysis using t-tests revealed significant differences (P < 0.001) between the PPL distributions of matched and mismatched pairs (Fig. 1e). The consistently higher PPL values observed in shuffled pairs indicate that our designed binders exhibit target specificity, as disrupting the intended target–binder pairing leads to predicted lower binding affinities.
We showcase two exemplary binder designs for mouse H-2Kb major histocompatibility complex (MHC) (Protein Data Bank (PDB) ID: 2OI9) and human tyrosine kinase (PDB ID: 1LCK) from our test set with detailed contact analysis (Fig. 1f). Analysis of the H-2Kb MHC (PDB ID: 2OI9) revealed that, despite high sequence identity (0.9) with the training set, PepMLM designed a distinct peptide (PSLGSVPYV) from the test binder (QLSPFPFDL) while maintaining equivalent binding quality (ipTM: 0.9). The designed peptide engages similar MHC binding residues, with terminal hydrophobic residues (proline and valine) serving as anchor points, suggesting similar biochemical properties to the experimental binder. In contrast, the tyrosine kinase complex (PDB ID: 1LCK) exhibited low sequence identity (0.26) with the training set, providing a stringent test of PepMLM’s capacity to design binders for novel targets. Here, the PepMLM-designed peptide (PPAEEIPP, ipTM: 0.82) exhibited more confident interaction compared to the test binder (EGQQPQPA, ipTM: 0.68). AlphaFold predictions demonstrated that both designed and experimental binders adopt similar spatial configurations and binding modes. Further analysis revealed that our designed peptide binders, despite having different sequences from the test binders, typically targeted the same binding pockets and displayed similar structural conformations, validating our language-model-based design approach (Supplementary Fig. 4 and Supplementary Table 3). In cases with lower predicted binding confidence scores, the designed binders exhibited distinct binding modes that appeared more optimal according to AlphaFold-Multimer predictions. However, it remains challenging to definitively ascertain whether our binders exhibit unique binding modes or if these observations are attributable to limitations in AlphaFold-based modeling8.
We further analyzed amino-acid-level patterns by generating 100 additional binders using both ESM-2 and PepMLM on the test set. At the amino acid composition level, PepMLM-designed sequences closely mirror the amino acid distribution of test binders, whereas ESM-2 exhibits strong biases toward serine (S), leucine (L) and randomly selected amino acids (X) (Fig. 1g), suggesting that PepMLM better captures the natural amino acid preferences in protein–peptide interactions after finetuning. More importantly, we conducted a detailed amino acids substitution analysis of generation, with particular attention to contact positions (defined using an 8-Å threshold). For each position in a test binder, we analyzed the amino acid types across corresponding positions in 100 designed binders. Overall, we observed 69.2% and 68.4% amino-acid-specific variations across all positions and contact positions, respectively. The diagonal elements of our generation matrix reveal that our model maintains a substantial probability of preserving the original amino acids in both contact and non-contact positions (Fig. 1h and Supplementary Fig. 5), indicating the ability of the mode to learn contextually appropriate amino acid choices given the target sequence. However, the model also demonstrates great versatility in generating diverse alternative amino acids. Within contact positions, we observed biochemically sensible generation patterns, including exchanges between hydrophobic residues (for example, valine to leucine at 13% and isoleucine to leucine at 17%) and similarly charged residues (lysine to arginine at 11% and aspartate to glutamate at 7.8%). Notably, positions containing cysteine exhibit highly conserved substitution patterns in our generation, potentially reflecting the critical role of cysteine in disulfide bond formation and structural stability. Together, these amino-acid-level analyses demonstrate PepMLM’s sophisticated understanding of biochemical properties and structural constraints in protein–peptide interactions at a fine-grained amino acid level.
Finally, to evaluate the generalizability of PepMLM beyond its training distribution, we analyzed the relationship between test target similarity to the training set and model performance (Supplementary Fig. 6). Notably, PepMLM consistently designed binders with low perplexity and high predicted binding affinity (ipTM), even for targets with less than 30% sequence identity, indicating that model performance does not rely on high homology and generalizes well to unseen protein substrates, motivating experimental characterization on diverse, disease-related targets.
PepMLM-derived peptides potently bind disease-implicated receptors in vitro
Our first goal was to establish the capacity of PepMLM to generate potent but specific peptide binders experimentally. To do this, we focused on two disease-related targets: neural cell adhesion molecule 1 (NCAM1), a key marker of acute myeloid leukemia29, and anti-Müllerian hormone type 2 receptor (AMHR2), a critical regulator of polycystic ovarian syndrome30. For both targets, four binders, each from PepMLM and RFdiffusion, were preliminarily screened using an ELISA (Supplementary Table 4), following which the most promising sequence from each algorithm (based on signal-to-noise ratio and maximum signal) was further characterized in triplicate. Results from initial screens indicate that all four PepMLM peptides for each target showed a binding response at concentrations as low as approximately 60 nM of AMHR2 or NCAM1-Fc fusions (Supplementary Fig. 8a,d). By contrast, RFdiffusion-generated peptides resulted in much poorer binders in both cases (Supplementary Fig. 8b,c). Although RFdiffusion produced moderate binders against NCAM1, binders generated against AMHR2 using the relevant crystal structure (PDB ID: 7L0J; chain B) showed minimal binding compared to blanks and BSA controls (Supplementary Fig. 8b,e). When preliminary screens were compared directly, we observed notably higher success rates of PepMLM-designed peptides in comparison to RFdiffusion peptides (Supplementary Fig. 8c,f). Further comparison of the best PepMLM and RFdiffusion binders against NCAM1 unveiled a clear difference in sensitivity and overall signal throughout all concentrations of NCAM1 tested (Fig. 2a and Supplementary Fig. 8g). Moreover, when probing the minimal concentration at which pMLM_NCAM1_2 can be distinguished from BSA controls, we observe significantly higher absorbance at approximately 30 nM (Fig. 2b; P = 0.0051). Notably, we show that none of the PepMLM peptides tested exhibited significant binding to BSA at concentrations up to 500 nM, that SUMO–polyglycine (polyG) constructs displayed no discernible difference in signal in the presence of either AMHR2 or NCAM1, and that use of generic IgG control as antigen yielded minimal signal up to 500 nM (Fig. 2a). Overall, these results corroborate our in silico observation that PepMLM generates promising binder candidates from target sequence alone with higher success rates than the current state-of-the-art binder design models.

Four NCAM1-targeting peptides were designed for both RFdiffusion and PepMLM as C-terminal SUMO-tag fusions and screened for potential binding to NCAM1 (Supplementary Fig. 8). a, Binding analysis of top candidate peptide against NCAM1 fused to C-terminal SUMO-tag as measured by ELISA. PepMLM-designed peptides were immobilized onto 96-well plates, incubated with serial dilutions of either NCAM1-Fc or BSA and subsequently detected with an anti-IgG–HRP conjugate and the signal was quantified by measuring the absorbance at 450 nm (A450). Data are represented as the mean ± s.e.m. (n = 3 biological replicates). b, Further ELISA binding analysis of pMLM_NCAM1_2 comparing successive serial dilutions of NCAM1-Fc to BSA control (200 nM) (n = 12 biological replicates) (P = 0.0051, P = 0.003, P < 0.0001, P < 0.0001). Box plots represent the mean value bounded by the IQR, with brackets representing minimum and maximum values. (Statistical significance was determined by an ordinary one-way ANOVA followed by a Dunnett’s multiple comparison test. Calculated P values are represented as follows: **P < 0.01; ***P < 0.001; ****P < 0.0001.).
PepMLM-derived uAbs degrade Huntington’s disease-related proteins in vitro
Having demonstrated PepMLM’s comparatively strong binder generation to RFdiffusion both in silico and in vitro, we next evaluated PepMLM peptides via fusion to E3 ubiquitin ligase catalytic domains by generating targeted uAbs to degrade pathogenic proteins in human cells (Fig. 3a)31. We focused our attention on Huntington’s disease, a monogenic dominant neurological disorder affecting more than one in 10,000 adults, caused primarily by an expanded CAG repeat in exon 1 of the HTT gene32. This results in an extended polyglutamine (polyQ) tract, leading to formation of the aggregation-prone mutant huntingtin protein (mHTT)33. Recently, it was shown that genetic knockdown of the mismatch repair-associated MSH3 protein reduces and inhibits mHTT repeat expansion34,35. We, thus, sought to degrade MSH3 at the posttranslational level via PepMLM peptide-guided uAbs.

a, Architecture and mechanism of uAb degradation system. CHIPΔTPR is fused to the C terminus of PepMLM-designed target-specific peptides and can thus tag endogenous target proteins for ubiquitin-mediated degradation in the proteasome following plasmid transfection. Created with BioRender. b, Quantitative immunofluorescence of RPE1 cells stained with anti-MSH3 antibody after transfection with plasmids encoding peptide–uAb or polyG–uAb control (n = 3 biological replicates, with >250 cells analyzed per replicate) (P = 1.00, P = 6.01 × 10−41, P = 5.60 × 10−44, P = 2.03 × 10−13, P = 1.28 × 10−55, P = 4.58 × 10−87) (statistical significance determined by one-sided Mann–Whitney U-test). Outliers were removed using the IQR method where values greater than the third quartile plus 1.5 times the IQR were excluded. Box plots represent the mean value bounded by the IQR, with brackets representing minimum and maximum values after outlier removal. c, Western blot analysis of TruHD-Q43/Q17M cell lysate in the presence or absence of doxycycline-dependent uAb expression. PolyG–uAb was used as a transfection ‘negative’ control, and vinculin was used as the loading control. d, Densitometry analysis of accompanying western blots of TruHD-Q43/Q17M cell lysate, normalized to polyG and vinculin controls (n = 3 biological replicates) (P = 0.0127, P = 0.0006, P = 0.0048, P = 0.0027, P = 0.0005). Data are represented as mean values ± s.e.m. (Statistical significance was determined by a paired one-tailed Student’s t-test. Calculated P values are represented as follows: **P < 0.01; ***P < 0.001.).
First, to design peptides for MSH3 degradation, we employed greedy decoding to determine the optimal binder length that yielded the lowest perplexity, followed by the generation of binders using top-k sampling, where k was fixed at 3 as previously described (Supplementary Table 4). After cloning these peptides into our uAb backbone and transfecting into genomically stable RPE1 cells, staining with fluorescently labeled anti-MSH3 antibody was used to quantify relative MSH3 levels. Quantitative immunofluorescence imaging revealed that five of six constructs tested (MSH3_pMLM_2–6) significantly reduced levels of MSH3 compared to polyG controls (Fig. 3b), demonstrating that anti-MSH3 PepMLM peptides are capable of robust degradation without extensive experimental screening.
We next sought to degrade the mHTT protein itself. To do this, we used TruHD fibroblasts, a genomically stable line that expresses the mHTT protein at a clinically relevant CAG repeat length of Q43 (ref. 36). As the line is heterozygous with both Q43/Q17 alleles, we designed PepMLM peptides targeting exon 1 with a polyQ repeat of 43. TruHD cell lines were then transfected with plasmids encoding for the five optimal candidates fused to a uAb domain under the control of a doxycycline-inducible promoter (Supplementary Table 4). HTT degradation was then measured with an anti-Huntingtin protein-specific antibody (EPR5526) recognizing the first 100 amino acids of HTT, which includes the polyQ region in both the presence and absence of doxycycline. Subsequent western blotting revealed minimal degradation prior to doxycycline induction, with visible ablation observed only after induction (Fig. 3c,d). Notably, all five candidates caused significant reduction of HTT levels compared to polyG control degradation (Fig. 3c,d). Taken together, the significant degradation of both MSH3 and HTT protein demonstrates the ability of PepMLM to design peptides that engage diverse, disease-related targets in cellulo.
PepMLM-derived uAbs degrade emergent viral phosphoproteins
Finally, we investigated whether PepMLM-derived uAbs could induce degradation of critical viral target proteins. As a key target class, we selected the viral phosphoprotein based on its relatively high sequence homology among strains of the selected viruses as well as its critical role in viral transcription and genome replication. Phosphoprotein sequences were selected for two emerging deadly viruses with high pandemic potential, the henipaviruses Nipah virus (NiV) and Hendra virus (HeV), both of which pose substantial threats to human health with recorded mortality rates of 50–100%37,38. A third phosphoprotein sequence was selected for the endemic virus human metapneumovirus (HMPV), whose infections occur more frequently than NiV and HeV, displaying seasonal cold-like symptoms that are severe and sometimes fatal in young children and elderly populations37. There are few to no vaccines or antiviral treatments approved for human use for these three viruses.
For each of the three viral phosphoproteins, 20 peptide-guided uAbs (Supplementary Table 4) were designed via PepMLM and then screened for their ability to induce proteasomal degradation of their respective phosphoproteins via western blotting and immunofluorescence imaging (Fig. 4). When uAbs were co-transfected with plasmids encoding for viral phosphoprotein target, a total of 37 degraders demonstrated between 20% and 49% average reduction in protein levels, suggesting an overall hit rate of approximately 63%, in strong agreement with our in silico hit rate shown in Fig. 1d (Fig. 4a–c). Although not significant, an obvious trend of diminished phosphoprotein levels can be observed with nearly all uAbs. Encouraged by these preliminary screening results, a smaller pool of candidate uAbs targeting HMPV were transfected into Vero AT cells, and, after infection, cells were stained using a fluorescent anti-HMPV phosphoprotein antibody. Immunofluorescent imaging shows near-complete amelioration of viral phosphoprotein levels for four uAbs (HMPV_12, HMPV_15, HMPV_18 and HMPV_19), which is likely to lead to reduced viral levels and infectivity in vivo (Fig. 4d).

a–c, Twenty uAb plasmids were co-transfected with plasmid DNA for each of the phosphoproteins from NiV (a), HeV (b) and HMPV (c) in HEK293T cells using PEI MAX. Whole-cell lysates were harvested 48 hours after transfection using RIPA buffer according to the manufacturer’s protocol. uAbs and phosphoproteins were probed using mouse anti-Flag and rabbit anti-HA antibodies, respectively, in addition to a mouse anti-β-actin loading control antibody. ‘EV’ is an empty pCAGGS vector, and ‘P’ is a phosphoprotein-only control. Quantification of reduced detection of target phosphoprotein was determined by densitometry as described in the Methods. Data are presented as mean values ± s.d. (n ≥ 3 biological replicates). d, Each immunofluorescent image shows Vero AT cells transfected with either an empty vector plasmid (pcDNA) or an HMPV uAb plasmid. Twenty-four hours after transfection, cells were infected with HMPV. Twenty-four hours after infection, ×10 immunofluorescent images were taken. HMPV phosphoprotein is shown in green, and cell nuclei are shown in blue. Each plasmid was tested with two biological replicates, each with two technical replicates.
Taken together, our experimental results strongly support the in silico benchmarking of PepMLM, with more than 60% of uAbs demonstrating moderate to strong degradation when PepMLM-designed peptides were used as modular guides (Figs. 3 and 4). Similar success was observed in binding analysis via ELISA, even compared to state-of-the-art structure-based binder design approaches (Fig. 2 and Supplementary Fig. 8). Activity of PepMLM-designed peptides, both in vitro and in cellulo, demonstrates the algorithm’s efficacy in binder design against diverse targets, in various cellular environments and in relevant contexts requiring degradation of mutant proteins, highly homologous viral proteins, and pathway-driving regulatory proteins. As an example of the latter, PepMLM-derived uAbs not only induce degradation of MESH1, an NADPH phosphatase that regulates ferroptosis, a form of cell death that is characterized by iron accumulation and lipid peroxidation39 (Supplementary Fig. 9a,b), but the top degrader, MESH1_pMLM_1, also inhibits ferroptosis-related cell death (Supplementary Fig. 9c). These results highlight the potential of PepMLM as a general-purpose platform for programmable peptide-guided modulation of protein function, enabling both binding and degradation across a wide spectrum of therapeutically relevant targets.
Discussion
Overall, PepMLM is a finetuned version of ESM-2 that employs a straightforward masking–unmasking scheme, offering an accessible framework for designing linear peptide binders. We acknowledge that PepMLM is not a standard generative sequence model, in comparison to more traditional autoregressive or discrete diffusion and flow matching models15,40,41,42,43,44. Despite our more minimalist formulation, we demonstrate that PepMLM yields strong binder designs, across in silico, in vitro and therapeutically relevant contexts. Our future work will build upon recent generative protein language models such as ProGen3 (ref. 45) or DPLM41,46 and incorporate more advanced sampling and search strategies tailored to encoder-only architectures. Such extensions will allow both binder generation and multi-objective conditioning for optimal therapeutic peptide design42,47.
To this point, we had used the lightweight ESM-2-650M model for PepMLM training, enabling flexible finetuning and inference. To assess the performance of larger models, we note that we additionally finetuned ESM-2-3B17 for peptide generation (PepMLM-3B) and evaluated it using the same methodology as employed for the ESM-2-650M version of PepMLM (PepMLM-650M). However, as illustrated in Supplementary Fig. 10, we did not observe a substantial improvement in either perplexity or hit rate for PepMLM-3B (36.02%). Considering the associated resource and inference costs, we provide our PepMLM-650M model as an accessible resource for effective linear peptide generation.
We envision that further improvements can be made to the PepMLM-650M model itself, enabling its adoption as a more universal tool for peptide binder design. For example, PepMLM can be retrained with modification-aware and variant-aware pLM embeddings to enable specificity to posttranslational isoforms over wild-type protein states46,48,49. Our future experimental work directions will include biochemical and molecular validation and characterization of the antiviral therapeutic potential of top selected uAbs within the groups tested for the emergent viral targets. We also plan to integrate PepMLM generation with high-throughput lentiviral screening to further evaluate its hit rate and input experimental data back into the algorithm, creating an active learning-based optimization loop50. As a note, we have not applied any experimental optimization of PepMLM-derived peptide binders, including further stabilization using cyclization or stapling—modifications that may improve therapeutic use47,51,52. We envision that through these additional developments, our accessible peptide generator, coupled with variants of our uAb and recent deubiquibody architecture53, will enable a CRISPR-analogous system to bind and modulate any target protein, whether structured or not.
Methods
Data curation
In the data curation phase, protein and peptide complexes were amalgamated from the PepNN and Propedia databases24,25. Initially, redundancy between the two datasets was eliminated, followed by the use of MMseqs2 to cluster the remaining protein sequences, setting a threshold of 0.8 (ref. 26). When protein sequences were identified within the same cluster and exhibited identical binder sequences, a single sequence was retained. This was followed by a manual filtering process, wherein protein sequences were sorted and those exhibiting high similarity (threshold of 80%) were removed to further mitigate homology issues. Consequently, a dataset comprising 10,203 entries was amassed, from which 10,000 were randomly allocated for training and 203 for testing. The maximum lengths for the binder and protein sequences were established at 50 and 500, respectively.
Conditional peptide modeling
Peptide binders are modeled in a distinctive manner, wherein the peptides are modeled conditionally based on the full protein sequence. Let p = (p1, p2, p3,…, pn) represent the target protein sequence of length n and b = (b1, b2, b3,…, bm) denote the binder of length m. The protein and peptide sequences are concatenated, incorporating special tokens of start, end and padding. Mask language modeling transforms this into a conditional modeling problem, where the objective is to reconstruct b given p and the entire masked b region. The entire model is updated with MLM loss, which can be represented as:
$${{\mathcal{L}}}_{\rm{{MLM}}}=-\frac{1}{m}\sum _{\mathrm{i} \in m}\log P\left({b}_{\rm{i}}{|p},{b}_{{\rm{mask}}}\right)$$
Through this methodology, the discrepancy between the designed binders and the ground truth is minimized, facilitating the approximation of the conditional probability, \(\mathop{\prod}\nolimits_{\rm{i=1}}^{m}P\left({b}_{\rm{i}}{|p}\right)\).
PepMLM training
The pretrained protein language model ESM-2 was used to facilitate full parameter finetuning. ESM-2, a transformer-based model, is adept at discerning co-evolutionary patterns across protein sequences. The concatenated protein and peptide sequences were tokenized at the amino acid level and input into the model. Deviating from the original training strategy of ESM, the entire binder sequence was exclusively masked, compelling the model to learn the relationship between the peptide binder and the protein. The ESM-2-650M and ESM-2-3B models were both trained for PepMLM. Both versions were trained on an NVIDIA 8x A100 640 GB DGX GPU system with PyTorch 2.01 and Python 3.10.10. Specific parameters are shown in Supplementary Table 1.
PepMLM generation
During the generation phase, the target protein sequence, along with a designated number of mask tokens (at end), was input into the model. Subsequently, the model greedily decodes logits at each masked position to identify peptide binders. To infuse greater diversity into the generation process, top-k sampling was implemented, wherein the model randomly selects the top k highest probability logits at each masked position.
PPL of PepMLM
The PPL of ESM-2 was adapted to focus specifically on the evaluation of peptide binder generation. Notably, the perplexity calculation is confined to the binder region or, in other words, the masked regions. Mathematically, the PPL is defined as:
$${\rm{Pseudo}}\mbox{-}{\rm{perplexity}}\left(b\right)=\exp \left\{-\frac{1}{m}\mathop{\sum }\limits_{\rm{i=1}}^{m}\log P\left({b}_{\rm{i}}|{b}_{j}\ne {\rm{i}},p\right)\right\}$$
In this equation, b represents the binder sequence, and m is the length of the binder sequence. This modification ensures a more focused evaluation of the designed peptide binders, aligning with the conditional modeling approach adopted in this study.
Peptide benchmarking
To assess the efficacy of the designed peptide binders, a benchmarking study was conducted on the test set. In the test set benchmarking, top-k sampling (k = 3) was employed to generate a single peptide binder for each target protein. Additionally, the original ESM-2 model was used to generate peptides, and random peptides of equivalent length were created. For ESM-2 generation, specifically, mask tokens of the same length were added at the end of target protein sequences for analogous model prediction and decoding as for PepMLM. The perplexity of the PepMLM was compared across four groups. PepMLM-designed binders and test binders were folded using AlphaFold2 ColabFold version 1.5.2, in conjunction with the protein sequences. Folding metrics including pLDDT and ipTM were gathered, which were used to correlate perplexity findings. For each test target protein, the ipTM scores of the test and designed binders were compared to determine the overall hit rate. Notice, as top-k sampling generates with randomness, the hit rate might vary or increase with different runs or k options.
RFdiffusion generation
In parallel to the PepMLM approach, RFdiffusion was employed to design peptide binders for both cases. For the given test set, RFdiffusion was tasked with generating one peptide binder per target protein, matching the length specified by the ground truth binders. The generated backbones were then converted into sequences using ProteinMPNN with initial guess and number of cycles of 3. The top sequence was selected via root mean square deviation. RFdiffusion inference code on ColabFold can be found at https://colab.research.google.com/github/sokrypton/ColabDesign/blob/v1.1.1/rf/examples/diffusion.ipynb.
Co-folding complex visualization
Structural visualization was performed using ChimeraX 1.7.1. The structures were superimposed using the MatchMaker tool, and interatomic contacts were identified using a van der Waals overlap threshold of ≥ −0.4 Å. The target proteins are shown in gray, whereas the PepMLM-designed and test binders are colored in red and blue, respectively. Their corresponding contact residues are highlighted in matching colors. Amino acid labels are displayed in the focused view.
For the rest visualization of AlphaFold-Multimer co-folding results from PepMLM-designed binder–protein complexes, an initial alignment with the corresponding test complex was performed using Biopython version 1.8.3, which facilitated a comparative visualization of selected complexes, encompassing both the designed and test binders. In these visualizations, the target protein was depicted in yellow, contrasting with the test and designed binders colored in blue and red, respectively. The visualizations were executed using py3Dmol version 2.0.4.
Alignment and identity
Target protein sequence similarity was assessed through two complementary approaches. Sequence identity between test and training sets was computed using the biotite Alignment method with an identity matrix. For each test target protein, the maximum identity score against all training set sequences was recorded. Additionally, a broader sequence similarity analysis was conducted using MMseqs2 (easy-search) to query both train and test target protein sequences against UniRef50, the training dataset of ESM-2.
Expression and purification of SUMO–peptide constructs
Peptides of interest were cloned into a pET-24a+ (Novagen) expression vector containing an N-terminal 6×-histidine–SUMO tag to facilitate downstream purification. Oligonucleotide primer pairs, each encoding for one half of the peptide sequences, were designed using NEBaseChanger V2 (https://nebasechanger.neb.com/) and then incorporated into the plasmid using Q5 site-directed mutagenesis, as per the manufacturer’s instructions. Site-directed mutagenesis reactions were carried out according to the same protocol. Plasmid assembly was verified using Sanger sequencing (GENEWIZ) and then transformed into chemically competent Escherichia coli BL21(DE3) cells. Starter cultures (3 ml of LB media, 50 µg ml−1 kanamycin) were inoculated from freshly streaked agar plates or glycerol stocks and grown at 37 °C with shaking at 225 r.p.m. overnight. Starter cultures were then diluted 1:500 in bulk cultures and grown to an optical density at 600 nm (OD600) of 0.6–0.8 and then induced at a concentration of 1 mM isopropyl β-d-thiogalactopyranoside (IPTG) overnight at 37 °C with shaking. Thirty minutes after induction, rifampicin was added to a final concentration of 150 µg ml−1. Cells were then collected by centrifugation (4,500g) at 4 °C and washed twice with ice-cold 1× PBS. The resulting cell pellets were frozen at −20 °C overnight, thawed to room temperature and then lysed using BugBuster protein extraction reagent (Millipore Sigma, 70584-3) supplemented with recombinant lysozyme (Millipore Sigma, 71110-3) and benzonase endonuclease (Millipore Sigma, E1014-25KU) for 20 minutes at room temperature with gentle rocking. The corresponding lysate was diluted with lysis buffer (1× PBS, 20 mM imidazole, 1× Halt protease inhibitor cocktail (Thermo Fisher Scientific, 78430)) and then centrifuged at 14,000g for 30 minutes. The cleared supernatant was mixed end over end at 4 °C for 30 minutes with HisPur Ni-NTA resin (Thermo Fisher Scientific, 88221) equilibrated with 20 mM imidazole in 1× PBS. Resin was centrifuged at 700g for 2 minutes and then washed three times with 50 mM imidazole in 1× PBS. Protein was eluted with three consecutive washes with 500 mM imidazole, concentrated (Millipore Sigma, 3K MWCO, UFC900308) and desalted using Zeba spin desalting columns (Thermo Fisher Scientific, 89892). Expression and purity of purified proteins in both the soluble and insoluble fraction, as well as purified fractions, were assessed using SDS-PAGE (Supplementary Fig. 8). Protein concentrations were quantified using a Qubit Protein Assay (Thermo Fisher Scientific, Q33211).
Sandwich ELISA
Purified SUMO-tagged peptide constructs were coated onto 96-well plates (Corning, CLS9018) at a concentration of 2 µg ml−1 in coating buffer (10 mM phosphate, pH 7.4) at a volume of 50–100 µl per well at 4 °C overnight with gentle rocking. Plates were washed once with Tris-buffered saline (50 mM Tris-HCl, 150 mM NaCl) supplemented with 0.05% Tween 20 (v/v) (TBS-T) and then blocked with 300 µl of SuperBlock in PBS (Thermo Fisher Scientific, 37516) per the manufacturer’s instructions. BSA, recombinant AMHR2-Fc (Sino Biological, 10673-H02H) and recombinant NCAM1-Fc (Sino Biological, 15785-H02H2) were serially diluted in triplicate or more in SuperBlock with 0.05% Tween 20, after which 100 µl of each solution was added to each well and incubated at room temperature with gentle rocking for 1.5 hours. Plates were then washed five times using 300 µl of TBS-T per well and then incubated with 100 µl of anti-human IgG (HRP) detection antibody (Thermo Fisher Scientific, A18805, diluted 1:10,000 in SuperBlock with 0.05% Tween 20) for 1 hour at room temperature. Plates were again washed five times with 300 µl of TBS-T and then incubated with 100 µl per well of 3,3′-5,5′-tetramethylbenzidine substrate (1-Step Ultra TMB-ELISA; Thermo Fisher Scientific, 34029) for 30 minutes at room temperature with gentle rocking. Finally, the reaction was quenched with 100 µl of 2 N H2SO4, and absorbance at 450 nm was immediately quantified using a Promega GloMax Discover plate reader.
Generation of mammalian plasmids
All uAb plasmids were generated from the standard pcDNA3 vector, harboring a cytomegalovirus promoter and a C-terminal P2A–GFP cassette as a transfection control. An Esp3I restriction site was introduced immediately upstream of the CHIPΔTPR coding sequence and flexible GSGSG linker via KLD Enzyme Mix (NEB) following polymerase chain reaction (PCR) amplification with mutagenic primers (GENEWIZ). For uAb assembly, PepMLM-derived peptide sequences (Supplementary Table 4) were human codon optimized for complementary oligo generation (GENEWIZ). Oligos were annealed and ligated via T4 DNA Ligase into the Esp3I-digested uAb backbone. Assembled constructs were transformed into 50 µl of NEB Turbo Competent E. coli and plated onto LB agar supplemented with the appropriate antibiotic for subsequent sequence verification of colonies and plasmid purification (GENEWIZ).
Sequences for human codon-optimized phosphoprotein genes for NiV (GenBank, AY029767), HeV (GenBank, MN062017) and HMPV (GenBank, AAS22075) were designed with HA tags on their N termini and flanked with restriction enzyme recognition sites for KpnI and XhoI on their 3′ and 5′ ends, respectively, for ligation into a mammalian pCAGGS vector.
Cell culture for target degradation
HEK293T and Vero AT cells were maintained in DMEM supplemented with 100 U ml−1 penicillin, 100 mg ml−1 streptomycin and 10% FBS. uAb-encoding plasmids (500 ng) were transfected into cells (4 × 105 per well in a 12-well plate) with Lipofectamine 2000 (Invitrogen) in Opti-MEM (Gibco). TruHD-Q43Q17M cells were maintained in Eagle’s Minimum Essential Medium with Earle’s Salts (EMEM) supplemented with 15% FBS, 1% NEAA (Gibco) and 1% GlutaMAX (Gibco). For HTT degradation studies, PepMLM peptides were transfected into fibroblasts using the SG cell line 4D-Nucleofector X Kit (Lonza). For viral protein degradation, transfections were done with HEK293T cells at approximately 90% confluency in six-well plates using a 4:1 µl:µg ratio of PEI MAX to DNA, following the transfection reagent manufacturer’s protocol. Target phosphoprotein plasmids were transfected at a 1:1 ratio with uAb plasmids for a total of 2 µg of DNA per well in Opti-MEM. Transfections were supplemented with Opti-MEM at approximately 5 hours after transfection. HMPV strain TN93-32 (BEI) was propagated in Vero AT cells for 5 days in DMEM supplemented with 100 U ml−1 penicillin, 100 mg ml−1 streptomycin and 2% FBS. RPE1 cells used for MSH3 studies were maintained in DMEM/Nutrient Mixture F-12 (Gibco) supplemented with 10% FBS and 10 µg ml−1 hygromycin B (Gibco) and transfected with PepMLM peptides using the TransIT-X2 Dynamic Delivery System (Mirus Bio).
MSH3 quantitative immunofluorescence
MSH3 antibody (Thermo Fisher Scientific, PA5-29829) was directly labeled using an Alexa Fluor Antibody Labeling Kit (Invitrogen). Transfected RPE1 cells were fixed 2 days after transfection using 4% paraformaldehyde (Thermo Fisher Scientific) for 20 minutes at room temperature and permeabilized using 0.2% Triton X-100 (BioShop) for 10 minutes at 4 °C. The cells were blocked using a blocking buffer (10% FBS in PBS) overnight and incubated with the labeled primary antibody diluted in blocking buffer (1:50) for 1 hour. The cells were then incubated with 2 µg ml−1 Hoechst 33258 (Thermo Fisher Scientific) for 5 minutes at room temperature. The imaging was done using an EVOS M7000 Imaging System (Thermo Fisher Scientific) at ×20. Cell segmentation and signal quantification was done using CellProfiler. When conducting downstream analysis, the TRITC signal from the PepMLM plasmid transfection was used to select for transfected cells. Data were analyzed to assess the statistical significance of differences in normalized intensities between the control group (polyG) and treatment groups (pMLM1–pMLM6). Outliers for all samples were removed using the interquartile range (IQR) method, where values greater than the third quartile plus 1.5 times the IQR were excluded to ensure robust comparisons. Statistical comparisons between the control and each treatment group were performed using a one-sided Mann–Whitney U-test. Significance thresholds were defined as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and *****P < 0.00001. Non-significant comparisons were denoted as ‘NS’. All analyses were conducted using Python, and plots were generated using Matplotlib.
HTT western blotting
The PepMLM peptide expression was induced using 1 µg ml−1 doxycycline (Sigma-Aldrich) 3 days before harvest and replenished every 2 days. On the day of harvest, TruHD-Q43Q17M cells were washed with 1× PBS and then lysed and scraped off using RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM EDTA) with protease and phosphatase inhibitors (Thermo Fisher Scientific) on ice. The mixture was incubated on ice for 5 minutes followed by centrifugation at 13,000 r.p.m. for 5 minutes at 4 °C. The supernatant was collected and quantified using a BCA Protein Assay Kit (Sigma-Aldrich). Then, 4× loading buffer (250 mM Tris pH 6.8, 40% glycerol, 8% SDS, 0.02% bromophenol blue) was added to the supernatant and incubated at 95 °C for 5 minutes. Immunoblotting was performed using precast 4–20% gradient gels (Bio-Rad) and then transferred onto an Immobilon-P PVDF membrane (Millipore). The membranes were blocked in 5% skim milk powder in 1× TBS-T (50 mM Tris-HCl, pH 7.5,150 mM NaCl, 0.1% Tween 20) at 4 °C overnight and then probed with rabbit anti-huntingtin antibody (Abcam, EPR5526, 1:5,000) or rabbit anti-vinculin antibody (Abcam, EPR8185, 1:5,000) in the same buffer for 1 hour at room temperature. The membranes were washed three times with 1× TBS-T and then three times with 2.5% skim milk powder in 1× TBS-T for 5 minutes each. The membranes were then probed with HRP-conjugated secondary antibodies (Abcam, 1:50,000) for 30 minutes at room temperature before being washed again and incubated with Immobilon Western Chemiluminescent HRP Substrate (Millipore) and imaged with a MicroChemi chemiluminescence detector (DNR Bio-Imaging Systems). Densitometry analysis was conducted using ImageJ. PolyG controls were first normalized by vinculin loading control with this normalized polyG being used to normalize uAb band degradation.
Viral phosphoprotein western blotting
HEK293T cells were harvested 48 hours after transfection and lysed using 1× RIPA buffer (Millipore) containing complete protease inhibitor (Sigma-Aldrich). The cells were incubated at 4 °C, rocking, for 40 minutes before being vortexed at 5-minute intervals for 20 minutes. Cell lysate supernatants were collected after centrifugation at 21,000g for 30 minutes at 4 °C. To denature samples for SDS-PAGE, cell lysates were mixed and incubated with 1.8% SDS containing 5% β-mercaptoethanol for 10 minutes at 95 °C before loading onto 10% acrylamide-Tris HCl gels. Proteins were separated at 100 V for 2 hours and then transferred onto 0.2-µm PVDF membranes at 0.5 A for 2 hours. Membranes were blocked in PBS with 0.2% Tween 20 (PBS-T) containing 4% BSA before staining in 1:1,000 dilutions of mouse anti-Flag (Millipore, F1804), mouse anti-β-actin (Santa Cruz Biotechnology, 47778) and rabbit anti-HA (BioLegend, 923502) primary antibodies. Secondary antibody staining was performed using 1:1,000 dilutions of goat anti-mouse Alexa Fluor 647 and goat anti-rabbit Alexa Fluor 488 secondary antibodies (Invitrogen, A21236 and A11008, respectively). Blocking, primary and secondary antibody membrane incubations were performed rocking at room temperature for 30 minutes, 1 hour and 30 minutes, respectively. Membranes were rinsed with PBS-T three times for 5 minutes after each antibody staining. All membranes were imaged using a Bio-Rad imager in respective Alexa Fluor channels. Densitometric quantification was performed using ImageLab for phosphoprotein and β-actin bands. Background densities from samples mock transfected with pCAGGS vector only were subtracted. Then, sample densities were normalized to their respective β-actin signals before normalization to their respective phosphoprotein controls. Data represent n ≥ 3 experimental replicates. Generation of bar graphs was performed using GraphPad Prism version 10, and the schematic diagram was made using BioRender (https://www.biorender.com/).
Immunofluorescent staining of viral phosphoprotein
Vero AT cells were seeded in 24-well plates to 90% confluency; after transfection and infection with HMPV, cells were washed with DPS twice and then fixed with 4% paraformaldehyde at room temperature to be subsequently permeabilized with a solution of 0.1% Triton X-100 in PBS. Custom polyclonal rabbit serum made against HMPV M was used for viral detection. After 1 hour, bound antibodies were detected with goat anti-rabbit secondary antibody conjugated with Alexa Fluor 488 (Invitrogen). Finally, the cellular nuclei were labeled with Hoechst (Thermo Fisher Scientific) in PBS for 10 minutes, and the images were examined using an ECHO Revolve microscope (BICO).
Cell culture for MESH1 degradation and ferroptosis protection
HEK293T cells were obtained from the Duke Cell Culture Facility and originated from the American Type Culture Collection (ATCC). The cells were cultured in DMEM 4.5 g l−1 glucose and 4 mM glutamine (Thermo Fisher Scientific, 11995-DMEM) and 10% heat-inactivated FBS (HyClone, SH30070.03HI) in a humidified incubator at 37 °C with 5% CO2. For MESH1 immunoblotting, HeLa cells originating from the ATCC were maintained in DMEM supplemented with 100 U ml−1 penicillin, 100 mg ml−1 streptomycin (Gibco) and 10% FBS. For uAb screening in reporter cell lines, 800 ng of pcDNA-uAb plasmids was transfected into cells in triplicate (3 × 105 per well in a 12-well plate) with Lipofectamine 2000 (Invitrogen) in Opti-MEM (Gibco). Cells were harvested 72 hours after transfection for subsequent immunoblotting.
MESH1 western blotting
On the day of harvest, cells were detached by adding 0.05% trypsin-EDTA and washing cell pellets twice with ice-cold 1× PBS. Cells were then lysed using a 1:100 dilution of protease inhibitor cocktail (Millipore Sigma) in Pierce RIPA buffer (Thermo Fisher Scientific). Specifically, the protease inhibitor cocktail–RIPA buffer solution was added to the cell pellet, and the mixture was placed at 4 °C for 30 minutes followed by centrifugation at 15,000 r.p.m. for 10 minutes at 4 °C. The supernatant was collected immediately to pre-chilled PCR tubes and quantified using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Then, 20 μg of lysed protein was mixed with 4× Bolt LDS Sample Buffer (Thermo Fisher Scientific) with 5% β-mercaptoethanol in a 3:1 ratio and subsequently incubated at 95 °C for 10 minutes prior to immunoblotting, which was performed according to standard protocols. In brief, samples were loaded at equal volumes into Bolt Bis-Tris Plus Mini Protein Gels (Thermo Fisher Scientific) and separated by electrophoresis. iBlot 2 Transfer Stacks (Invitrogen) were used for membrane blot transfer, and, after a 1-hour room-temperature incubation in 5% milk–TBS-T, proteins were probed with rabbit anti-HDDC3 antibody (Sigma-Aldrich, HPA040895, diluted 1:1,000) or rabbit anti-vinculin (Invitrogen, 700062, diluted 1:2,000) for overnight incubation at 4 °C. The blots were washed three times with 1× TBS-T for 10 minutes each and then probed with a secondary antibody, donkey anti-rabbit IgG (H + L) (HRP) (Abcam, ab7083, diluted 1:5,000), for 1 hour at room temperature. After three washes with 1× TBS-T for 10 minutes each, blots were detected by chemiluminescence using an Invitrogen iBright CL1500 Imaging System. Densitometry analysis of protein bands in immunoblots was performed using FIJI software as described at https://imagej.nih.gov/ij/docs/examples/dot-blot/. In brief, bands in each lane were grouped as a row or a horizontal ‘lane’ and quantified using FIJI’s gel analysis function. Intensity data for the uAb bands were first normalized to band intensity of either vinculin in each lane and then to the average band intensity for the polyG–uAb vector control cases across replicates.
Ferroptosis protection assay
HEK293T cells were reverse transfected using 1 μg of uAb plasmid and 3 μl of Mirus TransIT-LT-1 (Mirus Bio) transfection reagent for 48 hours, using the standard protocol for a 12-well plate as described by the manufacturer. The transfected HEK293T cells were transferred 2,500 cells per well to a 96-well plate, and 10 M erastin (Cayman Chemical) was added to the media to induce ferroptosis. Cell viability was measured 24 hours later using the Cell-Titer Glo (Promega) assay following the manufacturer’s protocol.
Statistical analysis and reproducibility
Unless otherwise noted, all data are reported as average values with error bars representing s.d. For samples performed in independent biological triplicates (n = 3) or more, statistical significance was determined by unpaired t-test, one-way ANOVA followed by a Dunnett’s multiple comparison test, paired one-sided Student’s t-test or Mann–Whitney U-test as indicated (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; *****P < 0.00001). Quantitative immunofluorescence sample sizes were 1,954, 1,311, 1,218, 3,662, 3,079, 2,887 and 3,845 for the polyG control and MSH3_pMLM_1–6, respectively. All graphs were generated using GraphPad Prism 10 version 14.4.1 or in Matplotlib. No data were excluded from the analyses unless specifically noted otherwise. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All source data, including raw and processed data (that is, raw immunoblots), have been deposited to the following Zenodo repository: https://doi.org/10.5281/zenodo.15122756 (ref. 54).
Code availability
All code and data used to train PepMLM are available at https://github.com/programmablebio/pepmlm. The PepMLM-650M datasets, code and model are also hosted on Hugging Face with an easy-to-use demo for peptide generation: https://huggingface.co/ChatterjeeLab/PepMLM-650M (ref. 55).
References
Chen, T., Hong, L., Yudistyra, V., Vincoff, S. & Chatterjee, P. Generative design of therapeutics that bind and modulate protein states. Curr. Opin. Biomed. Eng. 28, 100496 (2023).
Zhong, L. et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal. Transduct. Target. Ther. 6, 201 (2021).
Békés, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Dong, G., Ding, Y., He, S. & Sheng, C. Molecular glues for targeted protein degradation: from serendipity to rational discovery. J. Med. Chem. 64, 10606–10620 (2021).
Gao, H., Sun, X. & Rao, Y. PROTAC technology: opportunities and challenges. ACS Med. Chem. Lett. 11, 237–240 (2020).
Behan, F. M. et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568, 511–516 (2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023).
Gainza, P. et al. De novo design of protein interactions with learned surface fingerprints. Nature 617, 176–184 (2023).
Vaswani, A. et al. Attention is all you need. In Proc. 31st Conference on Neural Information Processing Systems 6000–6010 (NIPS, 2017).
Ofer, D., Brandes, N. & Linial, M. The language of proteins: NLP, machine learning & protein sequences. Comput. Struct. Biotechnol. J. 19, 1750–1758 (2021).
Elnaggar, A. et al. ProtTrans: toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2022).
Madani, A. et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 41, 1099–1106 (2023).
Ferruz, N., Schmidt, S. & Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 13, 4348 (2022).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Hie, B. L. et al. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol. 42, 275–283 (2023).
Brixi, G. et al. SaLT&PepPr is an interface-predicting language model for designing peptide-guided protein degraders. Commun. Biol. 6, 1081 (2023).
Bhat, S. et al. De novo design of peptide binders to conformationally diverse targets with contrastive language modeling. Sci. Adv. 11, eadr8638 (2025).
Portnoff, A. D., Stephens, E. A., Varner, J. D. & DeLisa, M. P. Ubiquibodies, synthetic E3 ubiquitin ligases endowed with unnatural substrate specificity for targeted protein silencing. J. Biol. Chem. 289, 7844–7855 (2014).
Chatterjee, P. et al. Targeted intracellular degradation of SARS-CoV-2 via computationally optimized peptide fusions. Commun. Biol. 3, 715 (2020).
Palepu, K. et al. Design of peptide-based protein degraders via contrastive deep learning. Preprint at bioRxiv https://doi.org/10.1101/2022.05.23.493169 (2022).
Abdin, O., Nim, S., Wen, H. & Kim, P. M. PepNN: a deep attention model for the identification of peptide binding sites. Commun. Biol. 5, 503 (2022).
Martins, P. et al. Propedia v2.3: a novel representation approach for the peptide-protein interaction database using graph-based structural signatures. Front. Bioinform. 3, 1103103 (2023).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. Preprint at bioRxiv https://doi.org/10.1101/2021.10.04.463034 (2021).
Johansson-Åkhe, I. & Wallner, B. Improving peptide-protein docking with AlphaFold-Multimer using forced sampling. Front. Bioinform. 2, 959160 (2022).
Sasca, D. et al. NCAM1 (CD56) promotes leukemogenesis and confers drug resistance in AML. Blood 133, 2305–2319 (2019).
di Clemente, N., Racine, C. & Rey, R. A. Anti-Müllerian hormone and polycystic ovary syndrome in women and its male equivalent. Biomedicines 10, 2506 (2022).
Zöllner, S. K. et al. Ewing sarcoma—diagnosis, treatment, clinical challenges and future perspectives. J. Clin. Med. Res. 10, 1685 (2021).
Ross, C. A. et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 10, 204–216 (2014).
Finkbeiner, S. Huntington’s disease. Cold Spring Harb. Perspect. Biol. 3, a007476 (2011).
Driscoll, R. et al. Dose-dependent reduction of somatic expansions but not Htt aggregates by di-valent siRNA-mediated silencing of MSH3 in HdhQ111 mice. Sci. Rep. 14, 2061 (2024).
O’Reilly, D. et al. Di-valent siRNA-mediated silencing of MSH3 blocks somatic repeat expansion in mouse models of Huntington’s disease. Mol. Ther. 31, 1661–1674 (2023).
Hung, C. L.-K. et al. A patient-derived cellular model for Huntington’s disease reveals phenotypes at clinically relevant CAG lengths. Mol. Biol. Cell 29, 2809–2820 (2018).
Gálvez, N. M. S. et al. Host components that modulate the disease caused by hMPV. Viruses 13, 519 (2021).
Gazal, S. et al. Nipah and Hendra viruses: deadly zoonotic paramyxoviruses with the potential to cause the next pandemic. Pathogens 11, 1419 (2022).
Ding, C.-K. C. et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat. Metab. 2, 270–277 (2020).
Alamdari, S. et al. Protein generation with evolutionary diffusion: sequence is all you need. Preprint at bioRxiv https://doi.org/10.1101/2023.09.11.556673 (2023).
Wang, X. et al. Diffusion language models are versatile protein learners. In Proc. 41st International Conference on Machine Learning 52309–52333 (ACM, 2024).
Chen, T., Zhang, Y., Tang, S. & Chatterjee, P. Multi-objective-guided discrete flow matching for controllable biological sequence design. Preprint at arXiv https://doi.org/10.48550/ARXIV.2505.07086 (2025).
Tang, S., Zhang, Y., Tong, A. & Chatterjee, P. Gumbel-Softmax Flow Matching with straight-through guidance for controllable biological sequence generation. Preprint at arXiv https://doi.org/10.48550/ARXIV.2503.17361 (2025).
Goel, S. et al. MeMDLM: de novo membrane protein design with masked discrete diffusion protein language models. Preprint at arXiv https://doi.org/10.48550/ARXIV.2410.16735 (2024).
Bhatnagar, A. et al. Scaling unlocks broader generation and deeper functional understanding of proteins. Preprint at bioRxiv https://doi.org/10.1101/2025.04.15.649055 (2025).
Vincoff, S., Davis, O., Tong, A., Bose, J. & Chatterjee, P. SOAPI: Siamese-guided generation of off-target-avoiding protein interactions. In ICLR 2025 Workshop on Generative and Experimental Perspectives for Biomolecular Design (2025).
Tang, S., Zhang, Y. & Chatterjee, P. PepTune: de novo generation of therapeutic peptides with multi-objective-guided discrete diffusion. In Proc. 42nd International Conference on Machine Learning (ICML, 2025).
Vincoff, S. et al. FusOn-pLM: a fusion oncoprotein-specific language model via adjusted rate masking. Nat. Commun. 16, 1436 (2025).
Peng, F. Z. et al. PTM-Mamba: a PTM-aware protein language model with bidirectional gated Mamba blocks. Nat. Methods 22, 945–949 (2025).
Zhao, L., Pal, A., Chen, T. & Chatterjee, P. A mammalian high-throughput assay to screen AI-designed protein degraders. In ICLR 2025 Workshop on Generative and Experimental Perspectives for Biomolecular Design (2025).
Vinogradov, A. A., Yin, Y. & Suga, H. Macrocyclic peptides as drug candidates: recent progress and remaining challenges. J. Am. Chem. Soc. 141, 4167–4181 (2019).
Moiola, M., Memeo, M. G. & Quadrelli, P. Stapled peptides—a useful improvement for peptide-based drugs. Molecules 24, 3654 (2019).
Hong, L. et al. Programmable protein stabilization with language model-derived peptide guides. Nat. Commun. 16, 3555 (2025).
Chen, T. et al. PepMLM: target sequence-conditioned generation of therapeutic peptide binders via span masked language modeling. Zenodo https://doi.org/10.5281/zenodo.15122756 (2025).
Chen, T. & Chatterjee, P. PepMLM: target sequence-conditioned generation of peptide binders via masked language modeling. Hugging Face https://doi.org/10.57967/hf/5858 (2025).
Acknowledgements
We thank the Duke Compute Cluster and Mark III Systems for providing database and compute resources that have contributed to the research reported in this paper. We also thank M. Ponnapati for curating initial peptide datasets and Y. Zhang for figure design discussion. Finally, we thank J. Chatterjee for emotional support during the preparation of this paper. The research was supported by institutional startup funds to the laboratory of P.C. from Duke University as well as funds from the CHDI Foundation, the Wallace H. Coulter Foundation, the Hartwell Foundation and National Institutes of Health (NIH) grants 3U54CA231630-01A1S4 and 1R21CA278468-01 to the laboratory of P.C. The work was also funded by institutional Cornell funds and NIH grant R01 AI109022 to the laboratory of H.C.A. and by the Krembil Foundation to the laboratory of R.T.
Ethics declarations
Competing interests
L.T.C. and P.C. are co-inventors on US patent applications 63/673,900 (‘Systems and methods for generating peptide binders via masked language modeling’) and 63/673,911 (‘Compositions for the targeted degradation of a protein and methods of making and using same’). P.C. and M.P.D. are co-founders of UbiquiTx, Inc., and are inventors of patents related to genetically encoded proteome editing technologies. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, L.T., Quinn, Z., Dumas, M. et al. Target sequence-conditioned design of peptide binders using masked language modeling. Nat Biotechnol (2025). https://doi.org/10.1038/s41587-025-02761-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41587-025-02761-2