
基于互作组的深度学习设计
作者:Wei, Dongqing
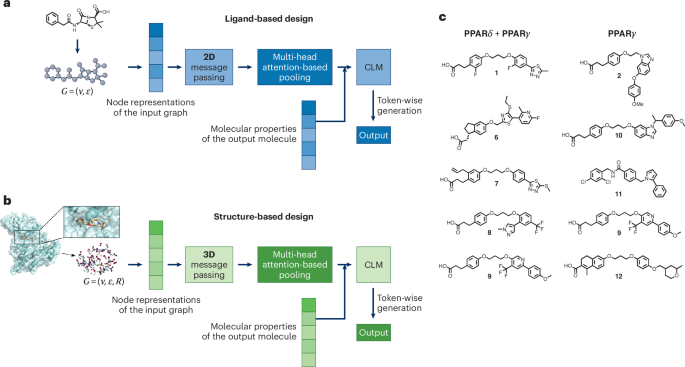
快速而准确地预测潜在的药物分子对于从头药物设计是一个相当大的挑战。一种基于相互作用组的深度学习方法已经被开发出来,其性能超过了标准的化学语言模型。
这是订阅内容的预览,通过您的机构访问
访问选项
访问Nature及Nature Portfolio其他54本期刊
购买Nature+,我们最具性价比的线上访问订阅套餐
24,99 €/ 30 天
随时取消
中国客户订阅信息
我们为中国客户专门设立了一个网站。请访问naturechina.com订阅此期刊。
购买此文章
- 在SpringerLink上购买
- 立即访问全文PDF
价格可能包括当地税款,该税款将在结账时计算。

参考文献
施耐德, G. & 费谢尔, U.自然·药物发现综述 4, 649–663 (2005).
袁,W. 等人。J. Chem. Inf. Model. 57, 875–882 (2017).
莫雷特,M. 等人。自然通讯 14, 114 (2023).
施耐德,P. 等人。自然·药物发现评论 19, 353–364 (2020).
阿茨 K. 等人。自然通讯 15, 3408 (2024).
梅嫩德斯,D. 等人。核酸研究杂志 47D930-D940 (2019).
伯杰, J. & 马勒, D. E.年度医学评论 53, 409–435 (2002).
阿布拉莫森,J. 等人。自然 630, 493–500 (2024).
格林希什尼科娃, D. Sci. 报告 11, 321 (2021).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E.国际机器学习大会. b, 1263–1272 (2017).
致谢
D.-Q.W.获得中华人民共和国国家重点研发计划政府间国际科技创新合作重点专项(2023YFE0199200)和国家自然科学基金(批准号32070662和32030063)的资助。
伦理声明
利益冲突
作者声明没有利益冲突。
关于本文文章

引用这篇文章
毛,X., 朱,Y. & 魏,D. 基于互作组的深度学习设计自然化学生物学(2024). https://doi.org/10.1038/s41589-024-01754-7
发布:
DOI: https://doi.org/10.1038/s41589-024-01754-7